
Synthesis and characterization of N-acyl-tetra-O-acyl glucosamine derivatives†
Abstract
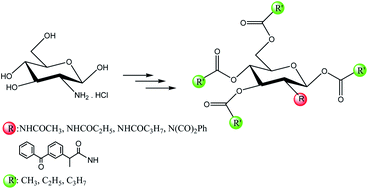
Novel 1,3,4,6-tetra-O-acyl-N-acyl-D-glucosamine derivatives were synthesized from glucosamine hydrochloride (GlcN·HCl) by the acylation with pyridine as a catalyst. A derivative of tetra-O-acetyl glucosamine contained ketoprofen, a non-steroidal anti-inflammatory drug (NSAID) with analgesic and antipyretic effects, was first synthesized. In analysis of the NMR spectra, the ratio of α:β-anomer showed that penta-acyl-D-glucosamine derivatives and N-acetylated glucosamines containing O-acyl groups have been only the α-anomer. Meanwhile, both the intermediates and the glucoconjugate compound of ketoprofen have only the β-anomer.
 Please wait while we load your content...
Please wait while we load your content...

