
High rate capacity retention of binder-free, tin oxide nanowire arrays using thin titania and alumina coatings†
Abstract
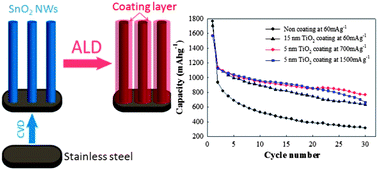
This paper reports the use of thin titania or alumina coatings on tin oxide nanowire arrays for high cyclability electrodes for lithium-ion batteries. We demonstrate that such coatings can significantly reduce irreversible capacity loss associated with the formation of a solid electrolyte interface and improve the capacity retention at high rates. Specifically, tin oxide nanowires grown on stainless steel substrates were conformally coated with thin films of either titania or alumina using atomic layer deposition and were tested as anodes in coin cells. Both titania and alumina coatings resulted in no initial capacity loss due to solid electrolyte interface formation in the first cycle. Tin oxide nanowire array electrodes coated with 5 nm thick titania layer and 1 nm thick alumina layer retained capacities of 767 and 725 mA h g−1 after 30 cycles using current density of 700 mA g−1. Both electrodes retained capacity around 664 mA h g−1 after 30 cycles using a current density of 1500 mA g−1, respectively. The results indicate that thin coatings acted as mechanical shells preserving the electrode nanostructure morphology necessary for high capacity retention. The study also showed that within the first two cycles, tin migrates out forming nanoclusters on the surface of nanowires due to both stress enhanced diffusion and the Kirkendall effect. The presence of tin nanoclusters on the surface of protective layers further enhances high rate capability.
 Please wait while we load your content...
Please wait while we load your content...

