
Glutaraldehyde in bio-catalysts design: a useful crosslinker and a versatile tool in enzyme immobilization
Abstract
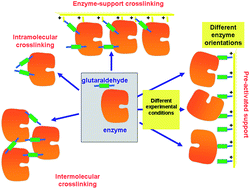
Glutaraldehyde is one of the most widely used reagents in the design of biocatalysts. It is a powerful crosslinker, able to react with itself, with the advantages that this may bring forth. In this review, we intend to give a general vision of its potential and the precautions that must be taken when using this effective reagent. First, the chemistry of the glutaraldehyde/amino reaction will be commented upon. This reaction is still not fully clarified, but it seems to be based on the formation of 6-membered heterocycles formed by 5 C and one O. Then, we will discuss the production of intra- and inter-molecular enzyme crosslinks (increasing enzyme rigidity or preventing subunit dissociation in multimeric enzymes). Special emphasis will be placed on the preparation of cross-linked enzyme aggregates (CLEAs), mainly in enzymes that have low density of surface reactive groups and, therefore, may be problematic to obtain a final solid catalyst. Next, we will comment on the uses of glutaraldehyde in enzymes previously immobilized on supports. First, the treatment of enzymes immobilized on supports that cannot react with glutaraldehyde (only inter and intramolecular cross-linkings will be possible) to prevent enzyme leakage and obtain some enzyme stabilization via cross-linking. Second, the cross-linking of enzymes adsorbed on aminated supports, where together with other reactions enzyme/support crosslinking is also possible; the enzyme is incorporated into the support. Finally, we will present the use of aminated supports preactivated with glutaraldehyde. Optimal glutaraldehyde modifications will be discussed in each specific case (one or two glutaraldehyde molecules for amino group in the support and/or the protein). Using preactivated supports, the heterofunctional nature of the supports will be highlighted, with the drawbacks and advantages that the heterofunctionality may have. Particular attention will be paid to the control of the first event that causes the immobilization depending on the experimental conditions to alter the enzyme orientation regarding the support surface. Thus, glutaraldehyde, an apparently old fashioned reactive, remains the most widely used and with broadest application possibilities among the compounds used for the design of biocatalyst.
- This article is part of the themed collection: Organic chemistry collection
 Please wait while we load your content...
Please wait while we load your content...

