
Carbon-induced Ru nanorod formation
Abstract
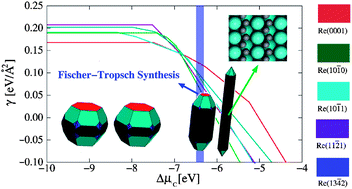
Using DFT calculations we study morphology of C-covered Ru nanostructures. We find that the equilibrium shape of clean Ru nanoparticles is a polyhedron consisting mainly of {0001}, {10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0}, and {101} faces. At low and intermediate coverages of C the shape does not deviate much from that of clean nanoparticles. However, at higher coverages the particle's morphology transforms from the polyhedron to a rod-like shape, which mainly consists of {100} faces. The formation of nanorods is due to a high stability of Ru(100), which is induced by the formation of chains of C atoms on this surface. We find that under conditions of Fischer–Tropsch synthesis, in which C forms from the hydrogenation of CO, the rod-like Ru nanoparticle is the most favorable shape.
0}, and {101} faces. At low and intermediate coverages of C the shape does not deviate much from that of clean nanoparticles. However, at higher coverages the particle's morphology transforms from the polyhedron to a rod-like shape, which mainly consists of {100} faces. The formation of nanorods is due to a high stability of Ru(100), which is induced by the formation of chains of C atoms on this surface. We find that under conditions of Fischer–Tropsch synthesis, in which C forms from the hydrogenation of CO, the rod-like Ru nanoparticle is the most favorable shape.
 Please wait while we load your content...
Please wait while we load your content...

