
Enantioselective synthesis of pyrazolone derivatives catalysed by a chiral squaramide catalyst†
Abstract
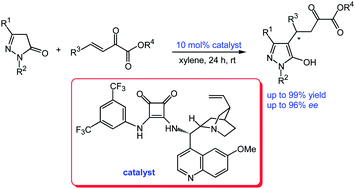
An efficient chiral squaramide-catalysed enantioselective Michael addition of pyrazolin-5-ones to β,γ-unsaturated α-ketoesters has been developed. The chiral pyrazolone derivatives were obtained in moderate to high yields (up to 99% yield) with high enantioselectivities (up to 96% ee) for most substrates. This catalytic asymmetric reaction provides valuable and easy access to chiral pyrazolone ketoester derivatives.
 Please wait while we load your content...
Please wait while we load your content...

