
From (+)-epigallocatechin gallate to a simplified synthetic analogue as a cytoadherence inhibitor for P. falciparum†
Abstract
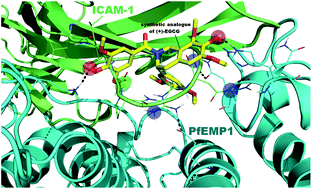
Parasite derived surface antigen PfEMP1 is a virulence factor of the human malaria parasite. PfEMP1 variants have been implicated in the cytoadherence of P. falciparum infected erythrocytes (iRBC) to several binding receptors on host vascular endothelium. Among them, binding to ICAM-1 seems to be related to severe manifestations of the disease such as cerebral malaria. The binding site for iRBC has been mapped to the BED-side of the N-terminal immunoglobulin-like domain of ICAM-1, and the DE-loop appears to be critical for binding. To date (+)-EGCG is the unique small molecule anti-cytoadherence inhibitor probably mimicking the DE-loop of ICAM-1. Here we report the discovery of a tetrahydroisoquinoline derivative, a prototype of a novel class of cytoadherence inhibitors, and an analogue of the natural compound characterized by a synthetically accessible scaffold. Molecular modeling analysis of (+)-EGCG and its synthetic tetrahydroisoquinoline analogue rationalized their binding mode to PfEMP1, confirming their ability to mimic the DE-loop.
 Please wait while we load your content...
Please wait while we load your content...

