Hydrothermal synthesis of Co3O4 with different morphologies towards efficient Li-ion storage†
Lingling Jinab,
Xiaowei Li*a,
Hai Mingab,
Haohe Wangab,
Zhenyong Jiaab,
Yu Fuab,
Jason Adkinsa,
Qun Zhoub and
Junwei Zheng*ab
aInstitute of Chemical Power Sources, Soochow University, Suzhou 215006, P. R. China. E-mail: jwzheng@suda.edu.cn; lixiaowei@suda.edu.cn; Fax: +86-512-67506056; Tel: +86-13013887911
bCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, P. R. China
First published on 19th December 2013
Abstract
In this study, Co3O4 with different morphologies (leaf, sheet, and cube) are successfully synthesized by a facile hydrothermal method followed by calcination treatment. Representative samples with different morphological structures are compared and evaluated as anode materials in lithium-ion batteries. Relative to the Co3O4-sheet and Co3O4-cube samples, the Co3O4-leaf samples exhibit excellent electrochemical performance with high storage capacity (1245 mA h g−1 after 40 cycles at 0.1 C) and superior rate capability (0.1, 0.2, 0.5, 1, and 2 C for 1028, 1085, 1095, 1038, and 820 mA h g−1, respectively); interestingly, the thinner the samples are, the better their performance. Moreover, assisted by characterization by cyclic voltammetry and electrochemical impedance spectroscopy, we draw a conclusion that the ultra-thin structures result in shorter path lengths for the transport of lithium ions and electrons, benefiting conductivity and fast charge–discharge rates. More importantly, for Co3O4, the respective structure's degree of thickness has a great effect on the electrochemical performance in lithium-ion batteries. This new concept might be extended to prepare other anode and cathode materials for advanced energy storage and conversion devices.
1. Introduction
Rechargeable lithium-ion batteries (LIBs) are one of the most promising candidates to become the major power source for mobile applications due to their excellent performance. However, current LIB technology is mainly based on graphite anode materials, which deliver a theoretical specific capacity of 372 mA h g−1, and these materials have a capacity which is far from meeting the growing demand for high capacity secondary batteries.1 Consequently, there is an urgent need to explore alternative anode materials with high capacity, good rate performance, and long cycle life. Recently, transition metal oxides (Co3O4, Fe3O4, NiO, CuO, etc.) have attracted an increasing amount of attention. Among them, Co3O4 is one of the most promising transition metal oxides due to its ability to react with up to 8 Li+ ions per formula unit, which gives a theoretical capacity of 890 mA h g−1, about three times that of graphite's.2–4 Even though Co3O4 possesses such high lithium storage capacity, the practical application of Co3O4 anode electrodes for LIBs is still largely restrained by the slow kinetics of the lithium ion and the electron transport within the electrodes as well as large volume changes observed over extended cycling.5,6A variety of attractive strategies has been utilized to solve these problems and can be mainly divided into two categories. One is the application of highly conductive carbon@Co3O4 composites, such as monosaccharide-derived carbon@Co3O4,7 CNTs@Co3O4,8 super-aligned carbon nanotube film@Co3O4,9 and onion-like carbon matrix@Co3O4 nanocomposites,10 which all exhibit a significantly improved performance in LIB applications compared to untreated Co3O4, since the highly conductive carbonaceous materials can increase the electronic conductivity and reduce damage from the volume expansion. The other strategy is to prepare Co3O4 with different morphologies, for example, hierarchical star-like Co3O4 micro/nanostructures,11 plate-like Co3O4 mesocrystals,12 sticktight-like structures, nanosheet Co3O4 particles,13 and needlelike Co3O4 nanotubes.14 Reported improvements have been made, in terms of the cycling stability and the rate performance of Co3O4, with the aforementioned morphologies and these improvements can be attributed to new techniques involving interconnected hierarchical nanostructures, use of small particle size with advantageous particle size distribution, as well as the use of porous and/or channel structured materials.
However, the effect of the respective structure's thickness on the electrochemical performance is not fully elucidated in previous studies which investigate the role of different morphologies of Co3O4 in LIBs. In some studies, Fe2O3 nanosheets,15 TiO2 thin film,16 SnO2 nanosheets,17 graphene nanosheets,18 NiO films,19 and LiMn2O4 thin films20 all show excellent electrochemical performance compared to that of their respective bulk material forms. Therefore, such kinds of ultra-thin structures probably could greatly facilitate lithium ion transport and increase electronic conductivity. Since these types of structures are also suitable for Co3O4, our conclusion provides significant guidelines for the development of LIBs.
At core of this objective, different morphologies of Co3O4 have been successfully prepared via a facile hydrothermal method and subsequent annealing in air under the same calcination conditions. Different morphologies of Co3O4 were investigated by scanning electron microscopy (SEM), in which we can see that the thickness of the Co3O4-leaf, Co3O4-sheet, and Co3O4-cube structures are about 30 nm, 100 nm, and 4 μm, respectively. Importantly, when these three samples were used as anode materials, the Co3O4-leaf samples exhibited the best cycling performance (1245 mA h g−1 after 40 cycles at 0.1 C) and good rate capacity (0.1, 0.2, 0.5, 1, and 2 C for 1028, 1085, 1095, 1038, and 820 mA h g−1, respectively) compared with Co3O4-sheet and Co3O4-cube samples. It is believed that such ultra-thin structures can facilitate lithium ion transport from the electrolyte to the electrode and increase the electronic conductivity of the electrodes, which is significant for the development of superior anode and cathode materials in LIBs.
2. Experimental section
2.1. Materials synthesis
2.2. Electrode fabrication
The electrochemical rate capability and cycling performance of samples were carried out with coin-shaped cells using a metallic lithium film as the counter electrode. The electrode was prepared by a coating method; slurries of 80 wt % active material, 10 wt % conducting carbon black (Super P), and 10 wt % polyvinylidene fluoride (PVDF) binder homogeneously mixed in N-methyl pyrrolidinone (NMP) were prepared into viscous slurries for efficient deposition. Afterwards, the slurries were stirred with a magnetic stirring apparatus until the powder mix was uniform. The slurries were coated onto the copper foil with an automatic film editor then dried in a vacuum oven at 120 °C for 12 h and cut into circular sheets. Then, the sheets were dried in a vacuum oven at 120 °C for 12 h again. The cells were assembled into CR2016 coin cells in a glove box filled with pure argon, in which the moisture and oxygen was strictly controlled to less than 1 ppm. Microporous polypropylene film (Celgard 2400) was used as the separator. The electrolyte was 1.0 mol L−1 LiPF6 in a mixture of diethyl carbonate (DEC) and ethylene carbonate (EC) in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w). The cells had a configuration of Li metal(−)|electrolyte|Co3O4(+), with a liquid electrolyte.
:1 (w/w). The cells had a configuration of Li metal(−)|electrolyte|Co3O4(+), with a liquid electrolyte.
2.3. Measurement and characterization
The obtained products were characterized by X-ray powder diffraction (XRD) using a X'Pert-ProMPD (Holand) D/max-γA X-ray diffractometer with Cu Kα radiation (λ = 0.154178 nm). The XRD measurement conditions were as follows: the scanning current was 40 mA, the scanning voltage was 40 kV. The SEM images were taken on a EVO 18 scanning electron microscope with an acceleration voltage of 20 kV. The N2 adsorption/desorption isotherms were obtained using a Quadrasorb 2MP (Micromeritics Instrument Corp.) Surface Area & Porosity Analyzer at 77 K. The specific surface area was calculated by the Brunauer–Emmett–Teller (BET) method. The pore size distribution was calculated by the Barrett–Joyner–Halenda (BJH) method.2.4. Electrochemical characterization
All the tests were carried out at room temperature.The electrochemical properties of products were tested in CR2016 coin cells. All the measurements were controlled and recorded automatically by the LAND CT2001A charge–discharge detector (China, Wuhan). Cyclic voltammetry (CV) tests were performed over the potential range of 0.01–3.00 V using a CHI1000C electrochemical workstation (Chenhua, Shanghai). Electrochemical impedance spectroscopy (EIS) was performed with a three-electrode system, using metallic lithium as the reference electrode. The applied frequency of the EIS test covered a range from 100 kHz to 10 mHz under an open circuit. The electrochemical analysis was tested using a Princeton Applied Research PARSTAT 2273 advanced electrochemical workstation system (AMETEK, America).
3. Results and discussion
3.1. Structural characterizations
The XRD patterns of as-synthesized precursors are first determined as shown in Fig. 1(a)–(c). It can be seen that Co3O4 phase (marked by *) is formed in all cases before heat treatment. There are other diffraction peaks for all of the samples. Those peaks could be assigned to their intermediates, such as 2CoCO3·3Co(OH)2·H2O,21 HCoO2,22 and CoCO3,23 respectively formed under different synthetic conditions, the detailed formation mechanism of the different morphologies are presented in the ESI.† After being annealed at 300 °C for 3 h in air, the crystallinity of the Co3O4 products increases and the peaks of intermediates disappear completely. All the diffraction peaks in Fig. 1(d)–(f) are in good agreement with the cubic phase of Co3O4 (JCPDS no. 42-1467), indicating that the intermediates were completely transformed to Co3O4 after calcination. | ||
| Fig. 1 XRD patterns of the precursors of (a) Co3O4-leaf, (b) Co3O4-sheet, (c) Co3O4-cube and the products of (d) Co3O4-leaf, (e) Co3O4-sheet, (f) Co3O4-cube. | ||
Fig. 2 depicts the SEM images of the precursors (Fig. 2a–c) and the Co3O4 products after heat treatment (Fig. 2d–f). These SEM images reveal that different morphologies of Co3O4 show good thermal stability since the overall structures are largely intact after calcination at 300 °C for 3 h, even though the surfaces of the samples are extraordinarily rough. It is clear that the morphology of Co3O4 crystal strongly depends on the coordination ligand and pH value of the media. The Co3O4-leaf can be formed only in the presence of urea in the pH = 9 media. As the pH value of the media is reduced to a pH of 8, Co3O4-cube is obtained. On the other hand, in the absence of urea, Co3O4-sheet is formed, at a pH of 9, in the media. The average dimensional parameters of the crystals with different morphologies are listed in Table 1. The thickness of the Co3O4-leaf and Co3O4-sheet are about 30 and 100 nm expressed from the SEM images (inset in Fig. 2d and e), respectively. The length of cube is about 4 μm at various dimensions.
 | ||
| Fig. 2 SEM images of the precursors of (a) Co3O4-leaf, (b) Co3O4-sheet, (c) Co3O4-cube and the products of (d) Co3O4-leaf, insert figure is the corresponding magnified SEM image. (e) Co3O4-sheet, insert figure is the corresponding magnified SEM image. (f) Co3O4-cube. | ||
| Morphology | Length | Width | Thickness |
|---|---|---|---|
| Co3O4-leaf | 4 μm | 1 μm | 30 nm |
| Co3O4-sheet | 1 μm | 1 μm | 100 nm |
| Co3O4-cube | 4 μm | 4 μm | 4 μm |
In addition to the sample size, surface area can be a factor as well; therefore, The N2 adsorption/desorption isotherms curves and the BJH pore size distribution plots obtained from the desorption branch are shown in Fig. S1.† The average parameters of the surface area and pore size with different morphologies are listed in Table S1.† The Co3O4-cube had the largest surface area; this was attributed to its multilayer structure which was composed of many stacked nanosheets. However, this confirmation of stacked nanosheets may not benefit the electrode–electrolyte contact and could lengthen the diffusion path of lithium ions and electrons, which have been proved by the following electrochemical performance test.
Different morphologies may bring about different electrochemical performance. The CV behaviors of Co3O4 products are evaluated to investigate Li-ion storage of each sample. Fig. 3 illustrates the CV curves of Co3O4-leaf, Co3O4-sheet, and Co3O4-cube for the first five cycles in the potential range of 0.01–3.0 V vs. Li/Li+ at room temperature at a scan rate of 0.1 mV s−1. It can be seen clearly that for all the samples, the CV curves of the first cycle are quite different from those of subsequent cycles, especially for the cathodic branch. For example, for the Co3O4-leaf, there are two well-defined peaks during the first cathodic scan (Fig. 3a). The weak peak at 1.16 V might be attributed to the formation of LixCo3O4.24 The strong peak at 0.85 V corresponded to the reduction of Co3+ and Co2+ to Co0 as well as a partially irreversible solid electrolyte interphase (SEI) layer and electrolyte decomposition.25 Two oxidation peaks at about 2.06 and 2.37 V, observed during the first anodic process, corresponds to the oxidation of Co0 to Co2+ and Co2+ to Co3+ accompanying the decomposition of Li2O.26,27 The corresponding electrochemical reactions can be described as follows:28
| Co3O4 + 2Li+ + 2e− ↔ Li2O + 3CoO | (1) |
| 3CoO + 6Li+ + 6e− ↔ 3Li2O + 3Co | (2) |
| Co3O4 + xLi ↔ LixCo3O4 | (3) |
| LixCo3O4 + (8 − x)Li ↔ 4Li2O + 3Co | (4) |
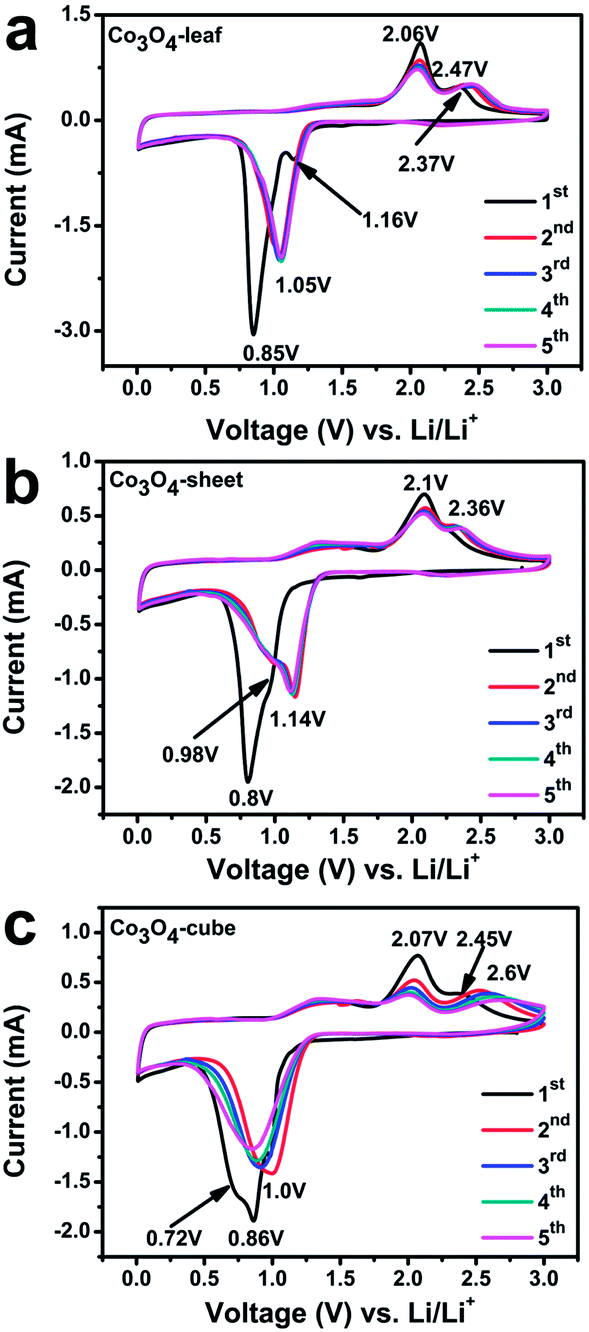 | ||
| Fig. 3 CV curves of (a) Co3O4-leaf, (b) Co3O4-sheet, (c) Co3O4-cube obtained at a voltage range of 0.01–3.0 V vs. Li/Li+ and potential scan rate of 0.1 mV s−1. | ||
A different CV behavior was observed during the second cycle, the CV curve is characterized by a distinct sharp cathodic peak of 1.05 V and two anodic peaks of 2.06 and 2.47 V. The potential shift of the reduction peak, relative to that in the first cycle, has been ascribed to the reduction of polarization effect, after the first cycle.12 Small shifts in the anodic peaks were also observed in Fig 3a. Moreover, in the next cycles, the CV curves of Co3O4-leaf sample overlap well, which may indicate a better stability of this material.
Fig. 3b and c show the CV curves of the Co3O4-sheet and Co3O4-cube. Unlike peaks in Fig. 3a, there are two over lapping cathodic peaks centered at 0.8 and 0.86 V for Co3O4-sheet and Co3O4-cube, respectively, in the first cathodic scan. The appearance of a “shoulder” peak at around 0.98 V for Co3O4-sheet could be attributed to the reduction of Co3+ to Co2+, while the “shoulder” peak at around 0.72 V for Co3O4-cube could be attributed to the reduction of Co2+ to Co0.29 In the second cycle, the cathodic peak shifts to 1.14 V and 1.0 V for Co3O4-sheet and Co3O4-cube, respectively, due to the polarization effect. Compare with the two reduction processes of the Co3O4-sheet and Co3O4-cube in Fig. 3b and c, the sharp reduction peak as well as the highest peak current in Fig. 3a facilitates the kinetic process of the electrochemical reaction, indicative a better electrochemical performance. And we can observe that the change on the cathodic peaks is more obvious than that on the anodic peaks. The possible reason maybe that Co3O4 is completely reduced to metallic cobalt which is well dispersed in the amorphous Li2O matrix after the first discharge process. So, the decomposition of Li2O is significant for the anodic process, but has little relationship to the thickness for as-prepared Co3O4 samples.29,30 Herein, the different electrochemical behaviors among three samples may result from their different morphologies. The ultra-thin structure of Co3O4-leaf provides a fast path transportation of lithium ions and electrons, resulting in better performance.
To better understand solid phase diffusion-controlled or surface-confined charge-transfer processes of the as-prepared sample, CV experiments at different scan rates (0.1, 0.2, 0.4, and 0.8 mV s−1) is conducted as given in Fig. 4. The peak currents of all the samples increase with increasing scan rate. The cathodic peaks shift to lower potential while the anodic peaks shift to higher potential as scan rate increases (Fig. 4a–c), showing increased electrochemical polarization. The linear dependence of the peak current (Ip) on the square root of the scan rate (v1/2) indicates an obvious diffusion-limited reaction in the cathodic process for the different morphologies of Co3O4.31 The relation is known as a typical Randles–Sevcik relation, which is expressed as: Ip = 0.4463n3/2F3/2CLiSR−1/2T−1/2DLi1/2v1/2, where n is the charge transfer number, F is the Faraday constant, CLi is the Li-ion concentration, S is the surface area of the electrode, R is the gas constant, and T is the absolute temperature (K), and all abovementioned parameters are constant.32 The variable, DLi is the diffusion coefficient, and the larger it is, the faster the lithium ions diffuse into the electrodes. As showed in Fig. 4d, the DLi of Co3O4-leaf samples is larger than that of the DLi of Co3O4-sheet and Co3O4-cube samples, indicating that the Co3O4-leaf samples with ultra-thin structure possess a fast lithium-ion diffusion ability, which is consistent with the CV results. We also checked the CV plots after 3 cycles (Fig. S2†), and we can see that the exact linear dependence remained. It was indicated that the lithium-ion diffusion ability among three samples didn't change after numbers of cycles.
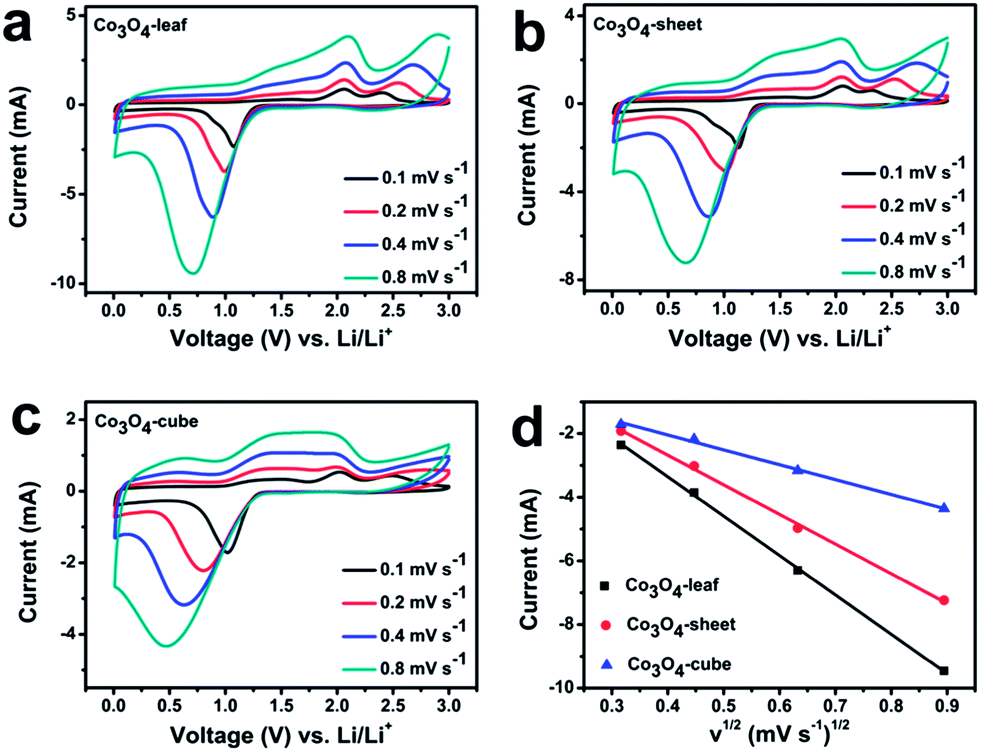 | ||
| Fig. 4 CV curves of (a) Co3O4-leaf (b) Co3O4-sheet (c) Co3O4-cube at scan rates ranging from 0.1 to 0.8 mV s−1. (d) The peak current vs. the square root of the scan rate. | ||
EIS is further used to characterize the electrochemical performance of the samples. Typically, EIS measurements are performed from 100 kHz to 10 mHz after 3 cycles in the fully charged state, the results are shown in Fig. 5. In the equivalent circuit (inset in Fig. 5), Rs is the ohmic resistance, Rf and CPE1 are associated with the resistance and the constant phase element of SEI film, Rct and CPE2 are associated with the charge-transfer resistance and the constant phase element of the electrode/electrolyte interface, Zw is associated with the Warburg impedance corresponding to the lithium-diffusion process.33,34 The Nyquist plots for the samples display a typical characteristic shape with the high frequency semicircle corresponding to the resistance Rf and CPE1 of the SEI film, the semicircle in the medium frequency region is assigned to the charge-transfer resistance Rct and CPE2 of the electrode/electrolyte interface. The inclined line corresponds to the lithium-diffusion process within the bulk of the electrode material.21,35 The fitted impedance parameters are listed in Table 2. It can be seen that the SEI film resistance Rf and charge-transfer resistance Rct of Co3O4-leaf electrode are 85.75 and 19.2 Ω, respectively, significantly lower than those of Co3O4-sheet (138.2 and 39.4 Ω) and Co3O4-cube (647.2 and 96.99 Ω). Smaller Rf value indicates its excellent film kinetics, as a steady SEI layer could be formed and well developed in fewer cycles. The lower Rct reflects the fact that charge transfer is facilitated on integral uniform surfaces of different morphologies of Co3O4 in the initial cycles, since Co3O4 itself is not electronically conductive. This fact confirms that the ultra-thin structure can preserve the high conductivity of the Co3O4-leaf electrode and greatly enhance rapid electron transport during the electrochemical lithium insertion/extraction reaction, resulting in significant improvement of electrochemical performances.
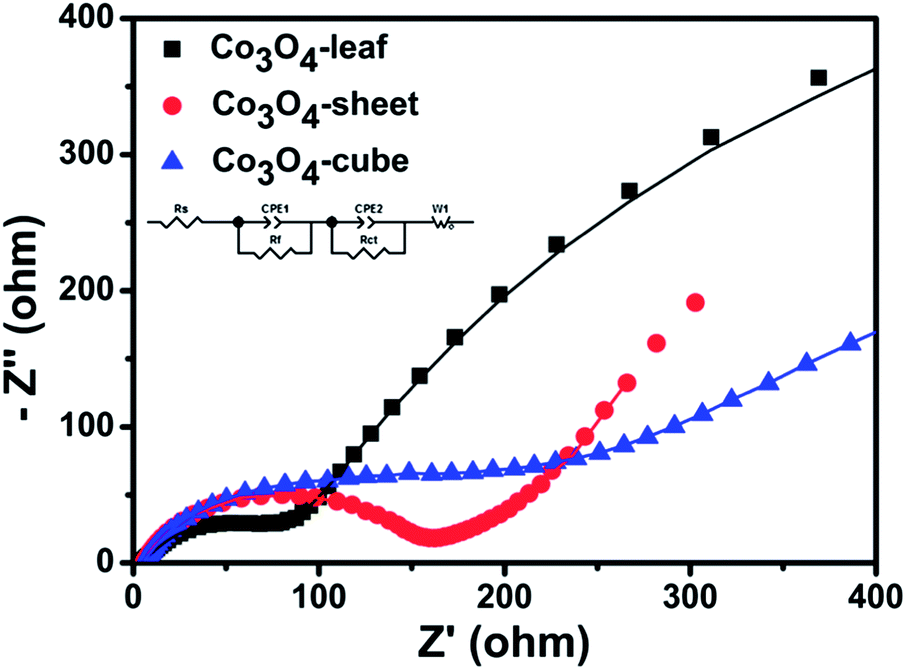 | ||
| Fig. 5 Nyquist plots of different morphologies of Co3O4 after 3 cycles in the frequency range of 100 kHz to 10 mHz, insert figure is the equivalent circuit. | ||
| Morphology | Rs (Ω) | Rf (Ω) | Rct (Ω) |
|---|---|---|---|
| Co3O4-leaf | 5.023 | 85.75 | 19.2 |
| Co3O4-sheet | 5.293 | 138.2 | 39.4 |
| Co3O4-cube | 2.826 | 647.2 | 96.99 |
Fig. 6 displays the voltage vs. capacity plots of Co3O4-leaf, Co3O4-sheet, and Co3O4-cube, respectively, at a rate of 0.1 C. Firstly, the Co3O4-leaf, Co3O4-sheet, and Co3O4-cube exhibit initial Li+ de-insertion capacities of 1092, 1443, and 1502 mA h g−1, which drastically reduced to 886, 1145, and 1000 mA h g−1 in the second cycle. The first cycle Columbic efficiency of Co3O4-leaf is calculated to be 78.4%, which is higher than that of Co3O4-sheet (74.2%) and Co3O4-cube (63.9%). This may result from the formation of the SEI film and further lithium consumption via interfacial reactions, which are due to the charge separation at the metal Co/Li2O phase boundary.36 Secondly, from the first discharge curves of the three samples, it can obviously be seen that there are two plateaus around 1.2 and 1.0 V for the Co3O4-leaf, but only one plateau at about 1.0 V for Co3O4-sheet and Co3O4-cube. This phenomenon is similar to that of the CV results. The ultra-thin structure of Co3O4-leaf is beneficial for electrolyte infiltration and accelerating lithium-ion diffusion, which leads to a complete chemical reaction and rapid response. Thirdly, after 10 cycles, the capacity of Co3O4-leaf and Co3O4-sheet is higher than the previous cycles which can be attributed to the gradual activation of nanocomposites. The capacity of Co3O4-cube in Fig. 6c decreases severely from the first cycle to the 10th cycle, which may be due to the large particle size of the Co3O4-cube with a longer electron transmission path.
 | ||
| Fig. 6 Voltage-Capacity curves of (a) Co3O4-leaf, (b) Co3O4-sheet and (c) Co3O4-cube cycles (1st, 2nd, 10th) between 0.01 and 3 V vs. Li/Li+ at a current rate of 0.1 C. | ||
To highlight the electrochemical performance of different morphologies of Co3O4, we test the cycling performance of the three electrodes at a current rate of 0.1 C and the results are compiled in Fig. 7a. The discharge and charge capacities for the first cycle are 1140 and 936 mA h g−1 for the Co3O4-leaf. The capacity exceed the theoretical capacity of Co3O4, indicating that the excess capacity is associated with increased charge storage within the polymeric surface layer.37–39 At the 30th cycle, the specific capacity rises to 1198 mA h g−1, while the capacity of the Co3O4-sheet is 1120 mA h g−1. For Co3O4-cube sample, the capacity fades rapidly from the first cycle to the 30th cycle. During all cycles, the Co3O4-leaf electrode presents much better electrochemical lithium storage performance and higher specific capacity than that of Co3O4-sheet and Co3O4-cube electrodes.
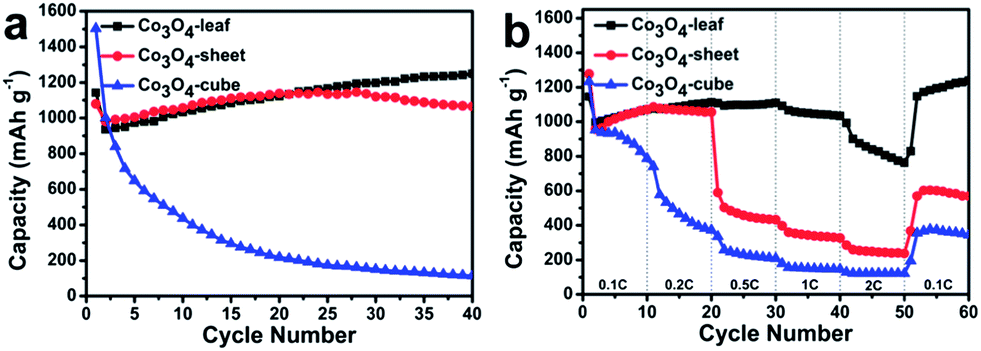 | ||
| Fig. 7 (a) Charge–discharge profiles of different morphologies of Co3O4 cycle performance at 0.1 C; (b) rate capability of different morphologies of Co3O4 at different current rates of 0.1 to 2 C. | ||
To further investigate the effect of the ultra-thin structure to the rate capability, the current rates are programmed to increase stepwise from 0.1 to 2 C (Fig. 7b). Among them, the Co3O4-leaf shows superior rate capability compared with the other two electrodes. The reversible capacities of Co3O4-leaf at 0.1, 0.2, 0.5, 1, 2, and 0.1 C are about 1023, 1075, 1096, 1048, 815, and 1178 mA h g−1, showing good stability of the structure. The capacity of Co3O4-sheet increases at 0.1 C and remains stable up to 0.2 C. A drastic capacity loss is observed at 0.5 C, which may due to the severe increase in electrical resistance and isolation.38 The capacity of Co3O4-cube decreased gradually at the current rate from 0.1 to 2 C, because of its larger and thicker size which lead to a long transportation path of lithium ions and electrons. The best rate capability of the Co3O4-leaf electrode are attributed to its ultra-thin structure. The ultra-thin structure not only decreased path lengths for the transport of electrons and lithium ions, it also improved electronic conductivity and accelerated ion diffusion rate. This conclusion is applicable even after the pulverization of Co3O4 samples. SEM images of three electrodes before and after 3 cycles are presented in Fig. S3.† It can be seen that Co3O4 samples pulverized into nanoparticles during cycling. However, the electrochemical performances of different morphologies were still different after the pulverization, and we can speculate that the contact between Co3O4 nanoparticles was concerned with the morphologies before the pulverization. For the ultrathin Co3O4-leaf samples, its internal nanoparticles and the surface nanoparticles were in contact with conductive additive even though after the pulverization. But for Co3O4-cube electrode, a large number of internal nanoparticles become inactive or less active since they can't contact with conductive additive (super P) efficiently after the pulverization, resulting these nanoparticles electronically disconnected to the current collector. So, it is reasonable to believe that the morphologies before cycling is significant to the electrochemical performance of the samples.
4. Conclusions
In summary, we implement a hydrothermal route to prepare the different morphologies of Co3O4 products followed by calcination at 300 °C for 3 h in air. It is found that the urea and the pH play a key role in controlling the formation of the different morphologies of Co3O4. When three samples are evaluated as anode materials, the Co3O4-leaf samples deliver the highest capacity of 1245 mA h g−1 after 40 cycles at 0.1 C; when the current rate increases to 0.1, 0.2, 0.5, 1, and 2 C, it still maintains a high capacity and good stability. The results confirm that the ultra-thin structure facilitates the diffusion of lithium ions and electrons, which makes it a promising anode material for next generation LIBs. Hopefully, this new concept will be extended to further research involving other anode and cathode materials in LIBs.Acknowledgements
Financial supports from the Nature Science Foundation of China (no. 20873089, 20975073), Nature Science Foundation of Jiangsu Province (no. BK2011272), Industry-Academia Cooperation Innovation Fund Projects of Jiangsu Province (no. BY2011130) and State Key Laboratory of Lithium Ion Battery Materials of Jiangsu Province are gratefully acknowledged.Notes and references
- X. Yao, X. Xin, Y. Zhang, J. Wang, Z. Liu and X. Xu, J. Alloys Compd., 2012, 521, 95–100 CrossRef CAS PubMed.
- Q. Hao, M. Li, S. Jia, X. Zhao and C. Xu, RSC Adv., 2013, 3, 7850 RSC.
- Y.-S. He, D.-W. Bai, X. Yang, J. Chen, X.-Z. Liao and Z.-F. Ma, Electrochem. Commun., 2010, 12, 570–573 CrossRef CAS PubMed.
- J. Wang, N. Yang, H. Tang, Z. Dong, Q. Jin, M. Yang, D. Kisailus, H. Zhao, Z. Tang and D. Wang, Angew. Chem., Int. Ed. Engl., 2013, 52, 6417–6420 CrossRef CAS PubMed.
- Z.-S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H.-M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS PubMed.
- Q. Q. Xiong, X. H. Xia, J. P. Tu, J. Chen, Y. Q. Zhang, D. Zhou, C. D. Gu and X. L. Wang, J. Power Sources, 2013, 240, 344–350 CrossRef CAS PubMed.
- N. Jayaprakash, W. D. Jones, S. S. Moganty and L. A. Archer, J. Power Sources, 2012, 200, 53–58 CrossRef CAS PubMed.
- S. M. Abbas, S. T. Hussain, S. Ali, N. Ahmad, N. Ali and K. S. Munawar, Electrochim. Acta, 2013, 105, 481–488 CrossRef CAS PubMed.
- X. He, Y. Wu, F. Zhao, J. Wang, K. Jiang and S. Fan, J. Mater. Chem. A, 2013, 1, 11121 CAS.
- Y. Wang, F. Yan, S. W. Liu, A. Y. S. Tan, H. Song, X. W. Sun and H. Y. Yang, J. Mater. Chem. A, 2013, 1, 5212 CAS.
- L. Li, K. H. Seng, Z. Chen, Z. Guo and H. K. Liu, Nanoscale, 2013, 5, 1922–1928 RSC.
- F. Wang, C. Lu, Y. Qin, C. Liang, M. Zhao, S. Yang, Z. Sun and X. Song, J. Power Sources, 2013, 235, 67–73 CrossRef CAS PubMed.
- X. Y. Feng, C. Shen, Y. Yu, S. Q. Wei and C. H. Chen, J. Power Sources, 2013, 230, 59–65 CrossRef CAS PubMed.
- X. W. Lou, D. Deng, J. Y. Lee, J. Feng and L. A. Archer, Adv. Mater., 2008, 20, 258–262 CrossRef CAS.
- M.-S. Wu, Y.-H. Ou and Y.-P. Lin, J. Electrochem. Soc., 2011, 158, A231 CrossRef CAS PubMed.
- K. F. Chiu, K. M. Lin, H. J. Leu, C. L. Chen and C. C. Lin, J. Electrochem. Soc., 2012, 159, A264–A268 CrossRef CAS PubMed.
- J. SongáChen, M. FengáNg and H. BináWu, CrystEngComm, 2012, 14, 5133–5136 RSC.
- P. Guo, H. Song and X. Chen, Electrochem. Commun., 2009, 11, 1320–1324 CrossRef CAS PubMed.
- X. Chen, N. Zhang and K. Sun, Electrochem. Commun., 2012, 20, 137–140 CrossRef CAS PubMed.
- Z. Quan, S. Ohguchi, M. Kawase, H. Tanimura and N. Sonoyama, J. Power Sources, 2013, 244, 375–381 CrossRef CAS PubMed.
- Y. Xiao, C. Hu and M. Cao, J. Power Sources, 2014, 247, 49–56 CrossRef CAS PubMed.
- Z. Chang, H. Li, H. Tang, X. Z. Yuan and H. Wang, Int. J. Hydrogen Energy, 2009, 34, 2435–2439 CrossRef CAS PubMed.
- S.-W. Bian and L. Zhu, RSC Adv., 2013, 3, 4212 RSC.
- X. H. Xia, J. P. Tu, J. Y. Xiang, X. H. Huang, X. L. Wang and X. B. Zhao, J. Power Sources, 2010, 195, 2014–2022 CrossRef CAS PubMed.
- L. Zhuo, Y. Wu, J. Ming, L. Wang, Y. Yu, X. Zhang and F. Zhao, J. Mater. Chem. A, 2013, 1, 1141 CAS.
- P. Shengjie and J. Mater, J. Mater. Chem. A, 2013, 1, 10935–10941 Search PubMed.
- G. Zhou, L. Li, Q. Zhang, N. Li and F. Li, Phys. Chem. Chem. Phys., 2013, 15, 5582–5587 RSC.
- M. V. Reddy, Z. Beichen, L. J. E. Nicholette, Z. Kaimeng and B. V. R. Chowdari, Electrochem. Solid-State Lett., 2011, 14, A79 CrossRef CAS PubMed.
- X. X. Zhang, Q. S. Xie, G. H. Yue, Y. Zhang, X. Q. Zhang, A. L. Lu and D. L. Peng, Electrochim. Acta, 2013, 111, 746–754 CrossRef CAS PubMed.
- G.-L. Xu, J.-T. Li, L. Huang, W. Lin and S.-G. Sun, Nano Energy, 2013, 2, 394–402 CrossRef CAS PubMed.
- C. Lai, Y. Y. Dou, X. Li and X. P. Gao, J. Power Sources, 2010, 195, 3676–3679 CrossRef CAS PubMed.
- J. Xie, T. Tanaka, N. Imanishi, T. Matsumura, A. Hirano, Y. Takeda and O. Yamamoto, J. Power Sources, 2008, 180, 576–581 CrossRef CAS PubMed.
- D. Fang, L. Li, C. Huang, C. Liang, Y. Ji, G. Li, G. Li, N. Wang, Z. Luo, W. Xu, J. Xu and L. Liu, J. Mater. Chem. A, 2013, 1, 13203–13208 CAS.
- B. Wang, G. Wang, Z. Zheng, H. Wang, J. Bai and J. Bai, Electrochim. Acta, 2013, 106, 235–243 CrossRef CAS PubMed.
- R. Ma, M. Wang, P.-P. Tao, Y. Wang, C. Cao, G. Shan, S. Yang, L. Xi, C. Y. Chung and Z. Lu, J. Mater. Chem. A, 2013, 1, 15060–15067 CAS.
- J. Chen, X.-H. Xia, J.-P. Tu, Q.-Q. Xiong, Y.-X. Yu, X.-L. Wang and C.-D. Gu, J. Mater. Chem., 2012, 22, 15056–15061 RSC.
- J. Zhu, L. Bai, Y. Sun, X. Zhang, Q. Li, B. Cao, W. Yan and Y. Xie, Nanoscale, 2013, 5, 5241–5246 RSC.
- G.-P. Kim, S. Park, I. Nam, J. Park and J. Yi, J. Mater. Chem. A, 2013, 1, 3872 CAS.
- J. Sun, H. Liu, X. Chen, D. G. Evans and W. Yang, Nanoscale, 2013, 5, 7564–7571 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra45904g |
| This journal is © The Royal Society of Chemistry 2014 |