
(Thio)urea-mediated benzoxazinone opening: mild approach towards synthesis of o-(substituted amido)benzamides†
Abstract
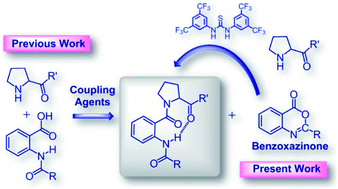
C-terminal activation of N-acylated o-aminobenzoic acids and their derivatives during coupling reactions with amines would often pose a challenge due to the formation of a benzoxazinone intermediate, which may resist reacting with amines. This communication reports a mild approach towards the installation of an amide bond via benzoxazinone ring opening utilizing Schreiner's (thio)urea organocatalyst.
 Please wait while we load your content...
Please wait while we load your content...

