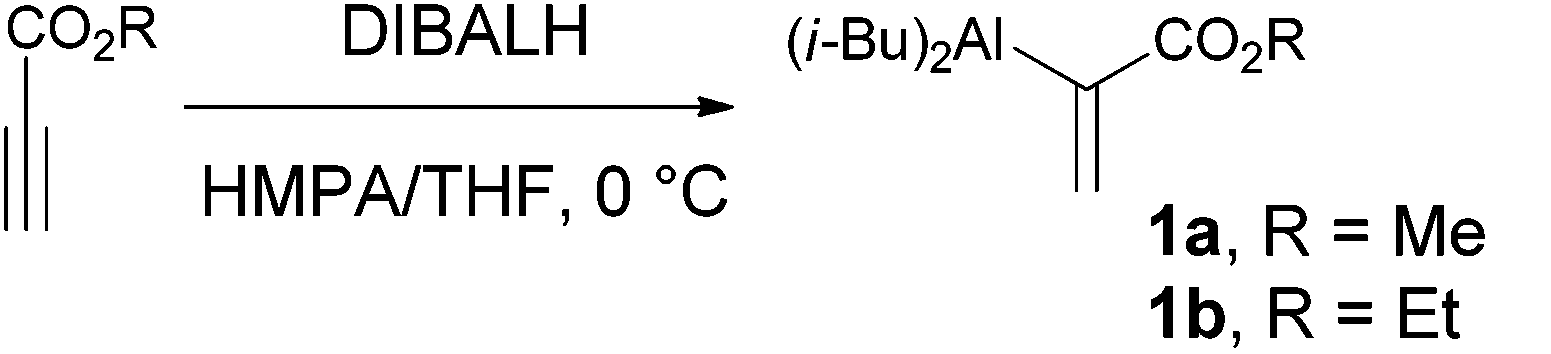DOI:
10.1039/C3RA45871G
(Paper)
RSC Adv., 2014,
4, 91-97
Diastereoselective vinylalumination for the synthesis of pericosine A, B and C†
Received
16th October 2013
, Accepted 29th October 2013
First published on
31st October 2013
Abstract
The vinylalumination of α-substituted aldehydes gave anti- and syn-adducts with moderate diastereoselectivity. The diastereomeric ratio was inverted by the addition of lithium or sodium perchlorates. Thus, both anti- and syn-adducts were isolated and transformed into the biologically active conduritols, pericosine B and C, respectively. Formal synthesis of pericosine A was achieved with the anti-adduct. The rationales for the different diastereoselectivity are discussed.
Introduction
[α-(Alkoxycarbonyl)vinyl]diisobutylaluminum (1), generated in situ from alkyl propiolate and diisobutylaluminum hydride in tetrahydrofuran/hexamethylphosphoramide (THF/HMPA), was introduced by Tsuda, Saegusa and colleagues in 1987 (eqn (1)).1 The reaction of the nucleophilic aluminium reagents 1 and aldehydes/ketones provided alkyl α-(1-hydroxyalkyl)acrylates, the same products furnished by the Morita–Baylis–Hillman reaction.2 This vinylalumination process circumvented the common drawbacks of the Morita–Baylis–Hillman reaction, such as slow reaction rates and the low reactivity of ketones and β-substituted acrylates. Vinylalumination is also applicable to other electrophiles, including allyl bromides,1a oxiranes and sulfinimines.3 Recently, Ramachandran's group improved the preparation of 1 by replacing the hazardous HMPA with environmentally benign 4-methylmorpholine N-oxide (NMO) increasing the applicability of this vinylalumination.4 Here, we report the application of this method to synthesize pericosine A, B and C (2a–c), which are cytotoxic metabolites of the sea hare-derived fungus Periconia byssoides OUPS-N133 reported in 1997 and 2007 by Numata and colleagues.5 It is interesting to note that both enantiomers of pericosine C were found in nature with a slight preference for the (−)-isomer.5b| | 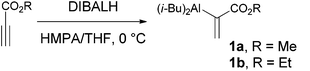 | (1) |
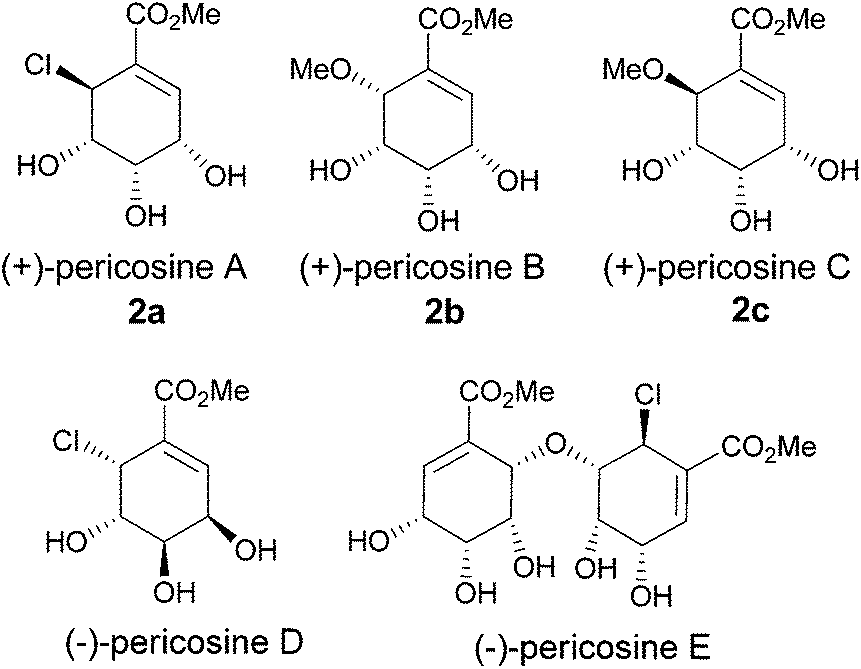
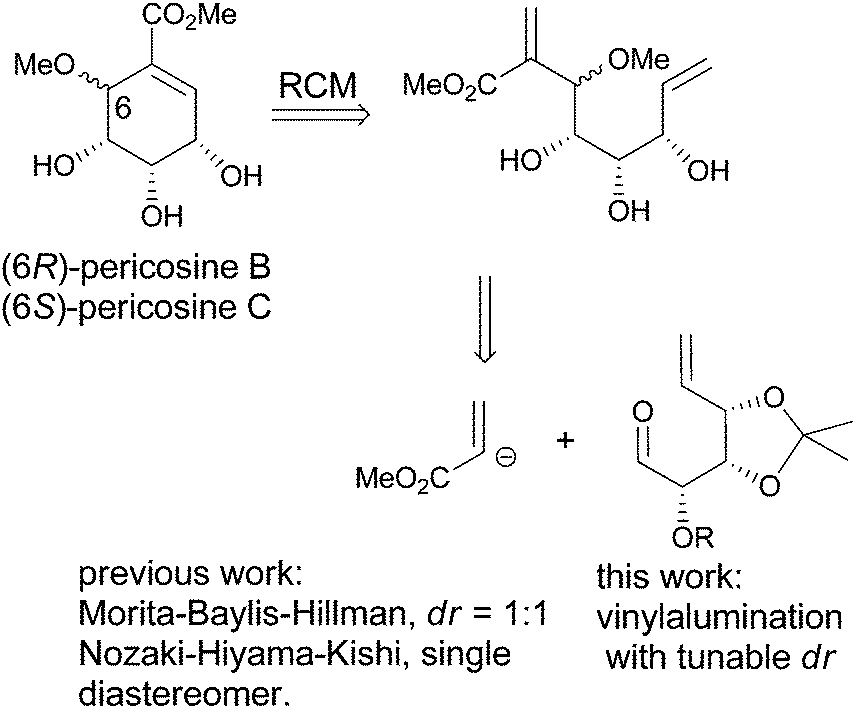
Pericosines belong to the family of conduritols and display a variety of biological activities including antitumour activity against some human cancer cell lines, inhibitory activity against epidermal growth factor receptor (EGFR), topoisomerase II and P388 leukaemia cells in mice;6 therefore, these compounds attract considerable attention from synthetic chemists (Fig. 1).7 Conventional approaches to access the six-membered cyclitol structure of pericosines include the use of shikimic acid, quinic acid and 3,5-cyclohexadiene-cis-1,2-diols prepared by the chemoenzymatic modification of substituted benzenes.8–10 Recently, both Chen's and Vankar's groups utilized ring-closing metathesis (RCM) to generate the cyclitol skeletons of pericosines, where the required dienes for RCM were prepared by the Nozaki–Hiyama–Kishi and Morita–Baylis–Hillman reactions, respectively (Scheme 1).11,12 We envisioned that the vinylalumination of the corresponding aldehydes could be a new method to synthesize the substrates for RCM, and the manipulation of the diastereoselectivity in this process could lead to the synthesis of both pericosine B and C. To our knowledge, the stereochemistry of the vinylalumination with α-substituted aldehydes has not been previously addressed.13
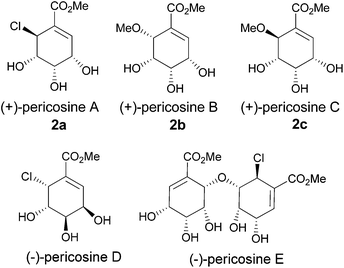 |
| | Fig. 1 Pericosine A–E. | |
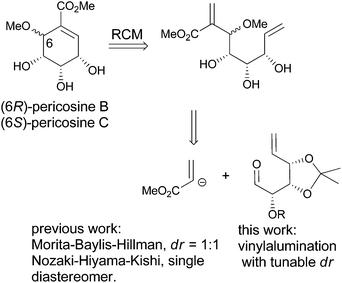 |
| | Scheme 1 Strategy for the synthesis of pericosines via RCM. | |
Results and discussion
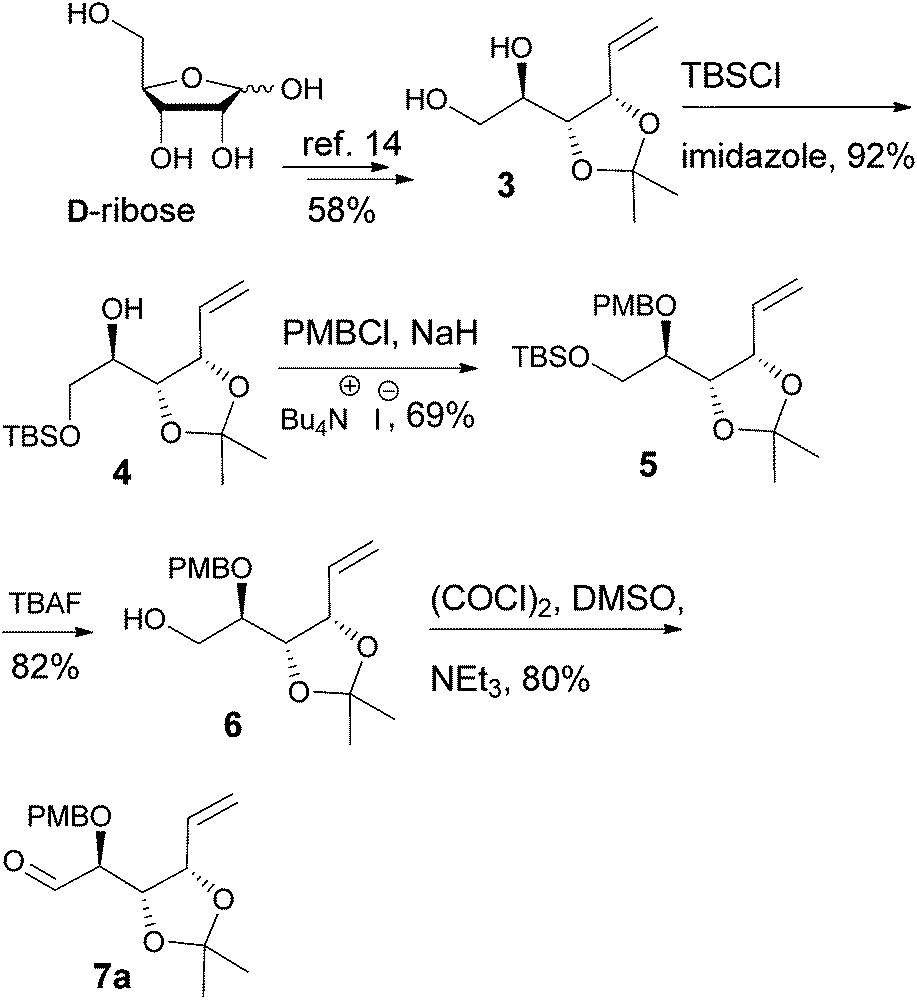
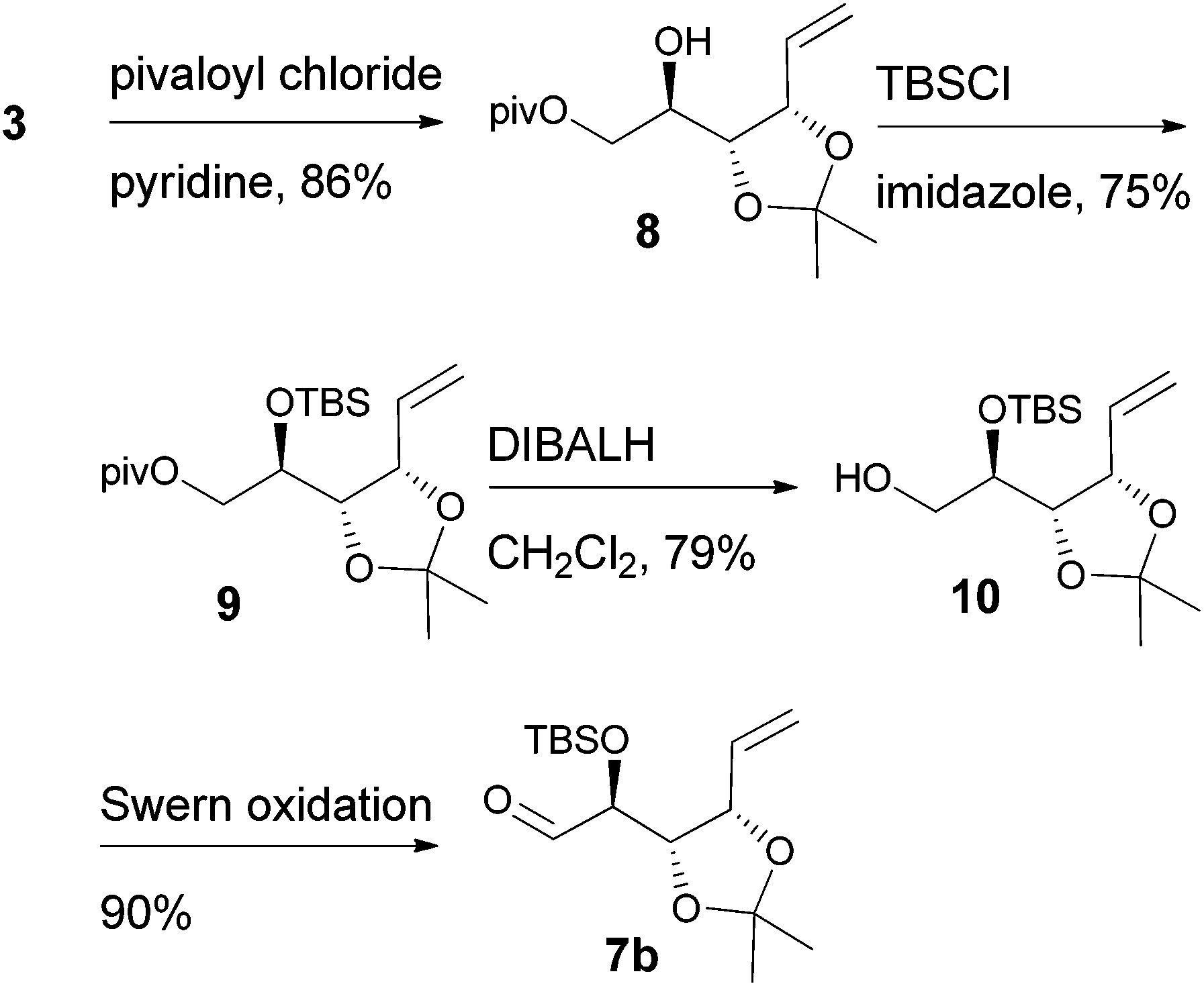
D-Ribose was converted to the diol 3 according to the reported procedures.14 The terminal hydroxyl group was differentiated by the formation of tert-butyldimethylsilyl ether 415 or pivalate 8. The remaining, secondary hydroxyl group was further protected as p-methoxybenzyl ether 5 or tert-butyldimethylsilyl ether 9, and the desired aldehydes 7a and 7b were prepared after the sequence of deprotection and Swern oxidation (Schemes 2 and 3).16
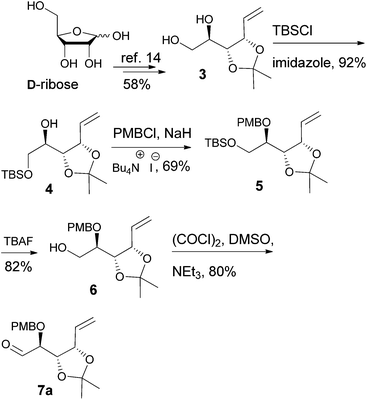 |
| | Scheme 2 | |
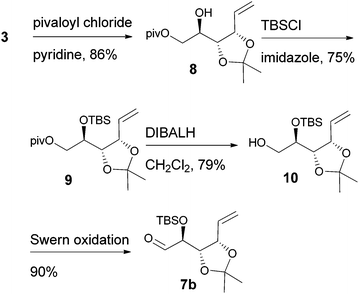 |
| | Scheme 3 | |
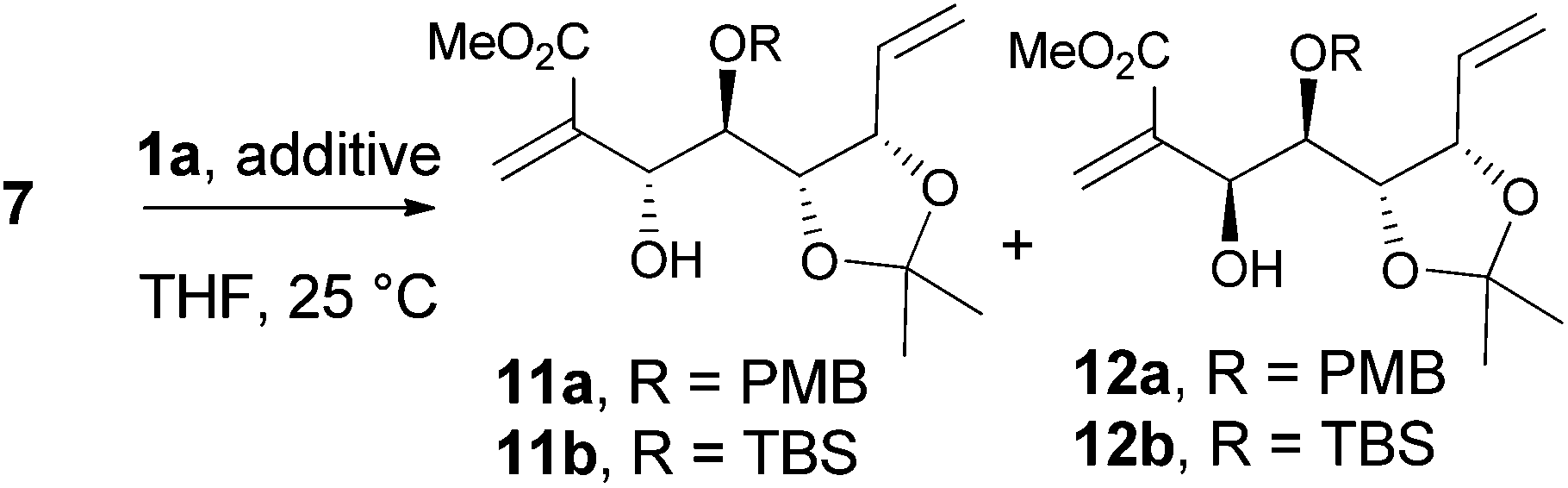
The vinylalumination of aldehyde 7 is summarized in Table 1. Consistent with previous reports, no product could be found when the additive was absent (entry 1).1a In the presence of HMPA, the reactions of 7a generated the two diastereomers 11a and 12a with a moderate diastereoselectivity (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and reasonable yields (entry 2). The two diastereomers were separable and isolated by column chromatography. Although our attempt to determine their stereochemistry by NMR analysis was inconclusive at this stage, this stereochemical outcome was later validated by further syntheses to pericosine B and C, respectively (vide infra). When the TBS-protected aldehyde 7b was used, the diastereoselectivity diminished to 1.2:1 (entry 3), this ratio is close to that observed in the Morita–Baylis–Hillman reaction (1:1) promoted by DABCO.11 Several Lewis acids were screened to improve the diastereoselectivity of this vinylalumination, and we found that alkali perchlorates had a strong influence on the reactions of 7a. As the equivalence of LiClO4 increased, the major product of the vinylalumination gradually shifted from 11a toward 12a (entries 4–6). The same trend was also observed when sodium perchlorate was applied (entries 7 and 8). The effect of KClO4 was negligible due to the much lower solubility of this salt in organic solvents (entry 9).17 The addition of LiClO4 has little effect on the vinylalumination of the TBS-protected aldehyde 7b (entry 10). Other Lewis acids, such as Mg(ClO4)2, Ca(ClO4)2, ZnBr2, MgBr2(OEt2) and AlMeCl2, did not generate the desired product, and the starting material 7a was recovered. The vinylalumination became very sluggish when a lower reaction temperature (0 °C) was applied.
:1) and reasonable yields (entry 2). The two diastereomers were separable and isolated by column chromatography. Although our attempt to determine their stereochemistry by NMR analysis was inconclusive at this stage, this stereochemical outcome was later validated by further syntheses to pericosine B and C, respectively (vide infra). When the TBS-protected aldehyde 7b was used, the diastereoselectivity diminished to 1.2:1 (entry 3), this ratio is close to that observed in the Morita–Baylis–Hillman reaction (1:1) promoted by DABCO.11 Several Lewis acids were screened to improve the diastereoselectivity of this vinylalumination, and we found that alkali perchlorates had a strong influence on the reactions of 7a. As the equivalence of LiClO4 increased, the major product of the vinylalumination gradually shifted from 11a toward 12a (entries 4–6). The same trend was also observed when sodium perchlorate was applied (entries 7 and 8). The effect of KClO4 was negligible due to the much lower solubility of this salt in organic solvents (entry 9).17 The addition of LiClO4 has little effect on the vinylalumination of the TBS-protected aldehyde 7b (entry 10). Other Lewis acids, such as Mg(ClO4)2, Ca(ClO4)2, ZnBr2, MgBr2(OEt2) and AlMeCl2, did not generate the desired product, and the starting material 7a was recovered. The vinylalumination became very sluggish when a lower reaction temperature (0 °C) was applied.
Table 1 Vinylalumination of aldehyde 7a
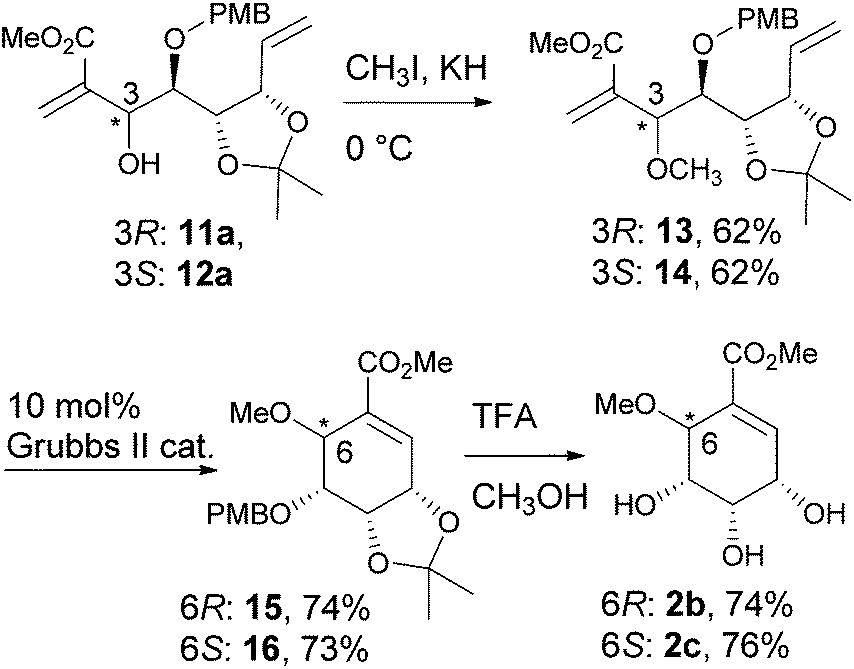
The vinylalumination adducts, 11a and 12a, were methylated to give 13 and 14, respectively. The following cyclisation by RCM and the removal of the acid labile protecting groups by TFA gave pericosine B (2b) and C (2c).18 The NMR spectra and optical rotation values of the synthetic 2b and 2c were consistent with the reported data,9b,5b which allowed us to confirm the stereochemical assignment of the vinylalumination adducts, 11a and 12a (Scheme 4).
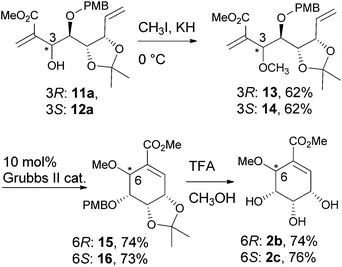 |
| | Scheme 4 Synthesis of pericosine B and C. | |
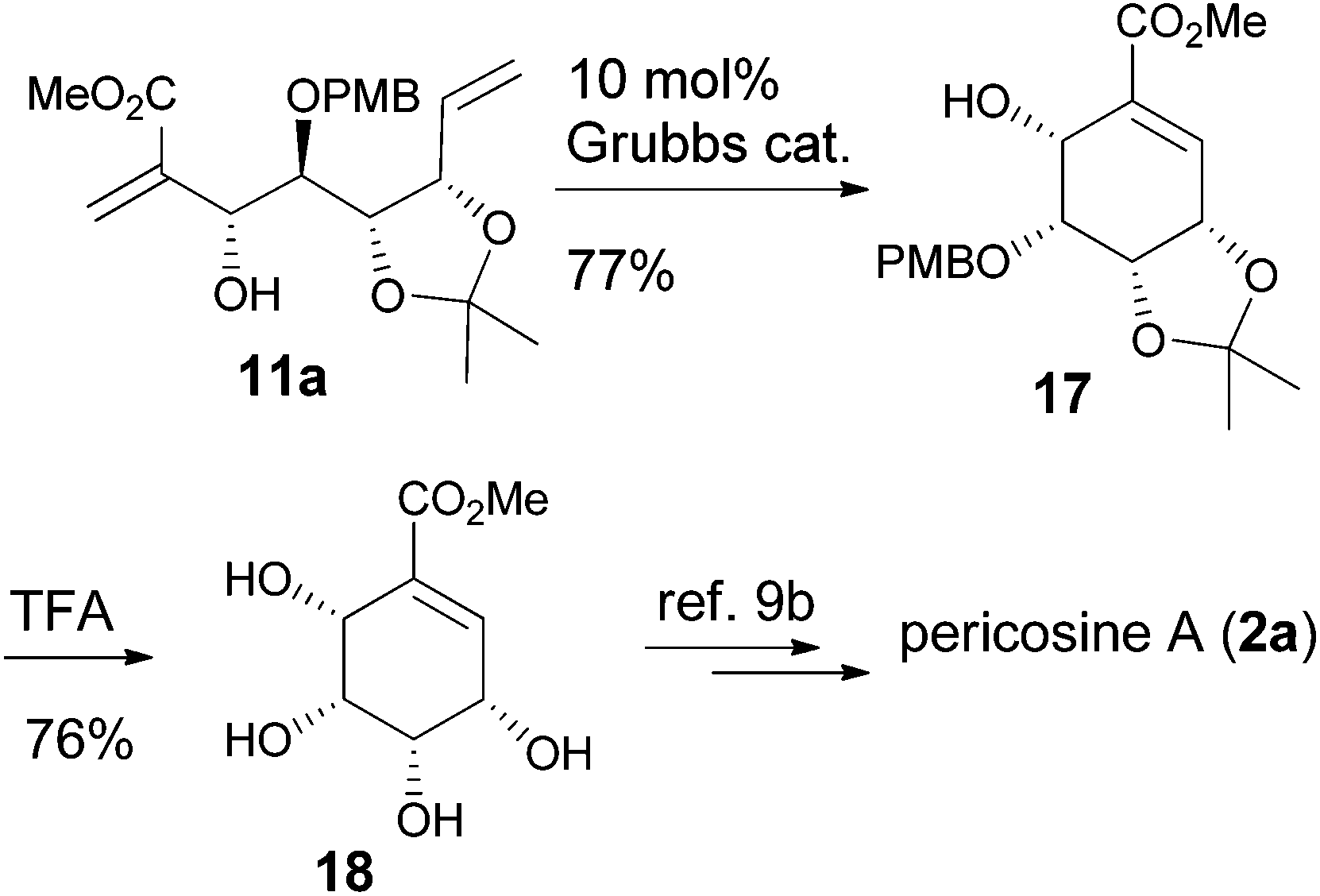
Direct RCM of 11a gave the cyclohexenyl alcohol 17, and the following deprotection provided the tetraol 18, which was the key intermediate for the synthesis of pericosine A by Stevenson's group (Scheme 5).9b
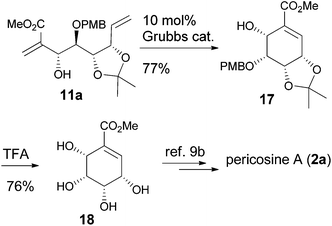 |
| | Scheme 5 Formal synthesis of pericosine A. | |
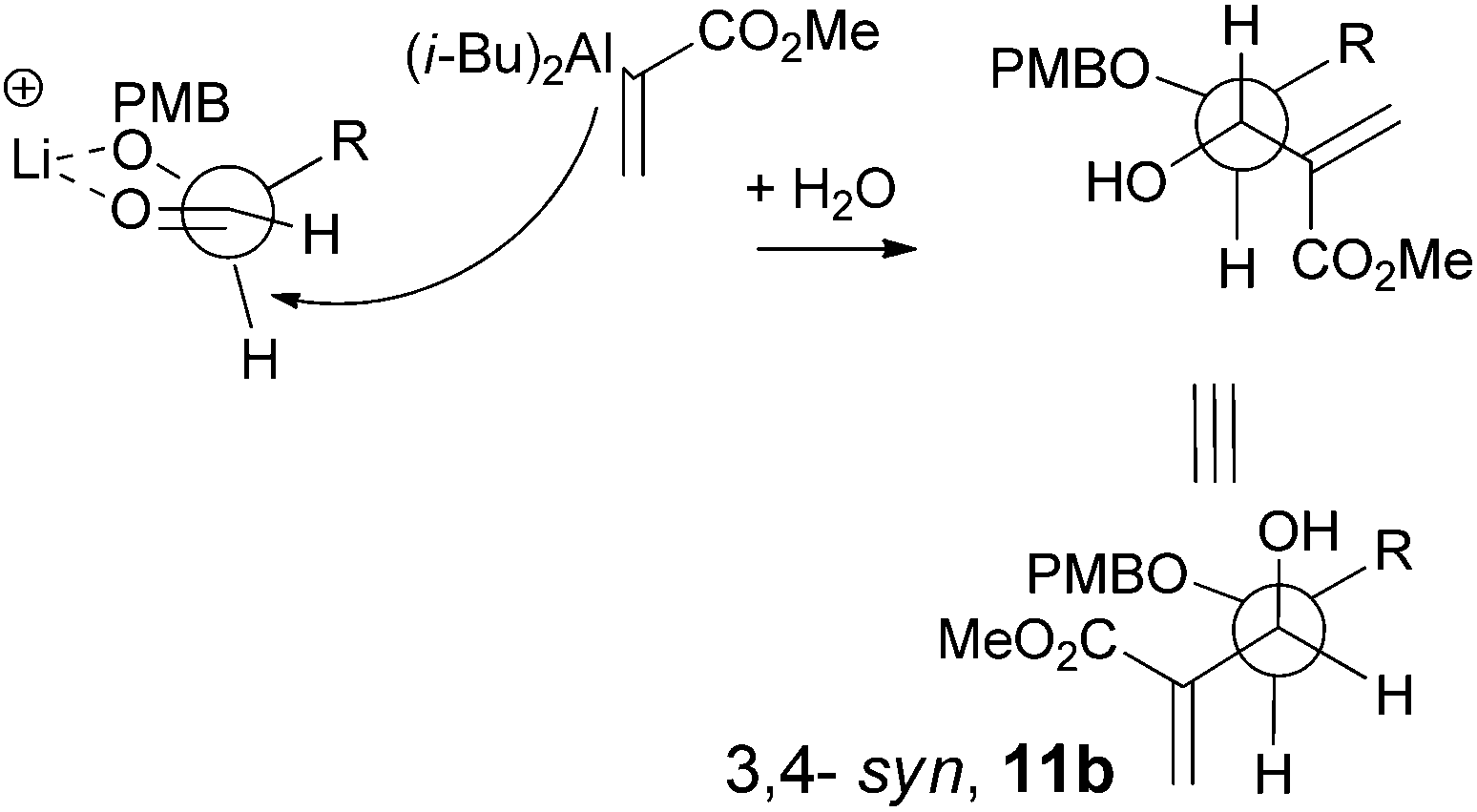
The stereochemistry of this vinylalumination merits further discussion. In the absence of LiClO4, the addition of 1 to 7a gave the 3,4-anti11a as the major product. This result is consistent with previous reports, where the addition of lithium enolate or allyl zinc to 2-O-benzyl-3,4-O-isopropylidene-erythrose, a truncated stereochemical analogue of 7a, also gave the anti-adducts as the major product,19 in accord with the Felkin–Ahn model.20 On the other hand, the formation of the 3,4-syn-adduct 12a from 7a is unusual.21 We propose that the excess amount of lithium or sodium cation enforced chelation control to generate the syn-adduct (Scheme 6) as the major product.22 However, the moderate diastereoselectivity (dr = 4) also suggested that the facial discrimination induced by this chelation control is mild. The insignificant effect of LiClO4 on the reaction of the highly sterically demanding, TBS-protected 7b supports the explanation of chelation control (Table 1, entries 3 and 10).23
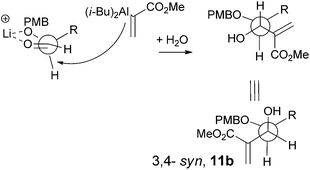 |
| | Scheme 6 Chelation control for the formation of 11b. | |
Conclusions
Diastereoselective vinylalumination using α-substituted aldehydes was achieved to provide α-methylene-β-hydroxy-γ-alkoxy-esters, such as 11 and 12. Therefore, the vinylalumination could be an alternative and stereoselective method to generate the same adducts as the Morita–Baylis–Hillman reaction. The addition of alkali perchlorates allowed us to manipulate the ratio of the two diastereomers (the 3,4-anti versus 3,4-syn) from the original 4:1 to 1:4. Three further steps transformed the anti- and syn-adducts to the conduritols, pericosine B and C, respectively. Formal synthesis of pericosine A was also achieved via RCM and deprotection of the anti-adduct. Further study utilizing this stereoselective vinylalumination for natural product synthesis is ongoing.
Experimental section
tert-Butyl((R)-2-((4S,5S)-2,2-dimethyl-5-vinyl-1,3-dioxolan-4-yl)-2-((4-methoxybenzyl)oxy)ethoxy)dimethylsilane (5)
A solution of 4 (2.0 g, 6.62 mmol), 4-methoxybenzyl chloride (2.07 g, 13.24 mmol) and THF (5 mL) was added to the suspension of sodium hydride (0.63 g, 26.47 mmol) and THF (20 mL) 0 °C. The reaction mixture was stirred at rt for 20 h, quenched with sat. NH4Cl(aq) (2 mL), concentrated, diluted with water (30 mL) and extracted with ethyl acetate (25 mL × 3). The organic layers were combined, dried over Na2SO4, filtered and concentrated. The crude product was further purified by column chromatography (SiO2, EtOAc–hexanes, 1:12; Rf 0.50) to give 5 (1.92 g, 4.56 mmol, 69%) as a colourless oil. [α]D20 −21.9 (c 0.69, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 0.07 (s, 6H), 0.91 (s, 9H), 1.33 (s, 3H), 1.46 (s, 3H), 3.46–3.52 (m, 1H), 3.71–3.77 (m, 1H), 3.78 (s, 3H), 3.99 (d, J = 11.1 Hz, 1H), 4.16–4.22 (m, 1H), 4.37 (d, J = 10.6 Hz, 1H), 4.65 (dt, J = 6.4 Hz, J = 0.9 Hz, 1H), 4.75 (d, J = 10.6 Hz, 1H), 5.22 (dd, J = 10.6 Hz, J = 1.5 Hz, 1H), 5.36 (dd, J = 17.1 Hz, J = 1.5 Hz, 1H), 5.93 (ddd, J = 6.7 Hz, J = 10.4 Hz, J = 18.2 Hz, 1H), 6.84 (d, J = 8.3 Hz, 2H), 7.23 (d, J = 8.3 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 159.1, 134.5, 130.7, 129.3, 117.0, 113.7, 108.4, 78.7, 78.2, 76.6, 71.7, 63.8, 55.2, 27.8, 25.9, 25.3, 18.3, −5.4; IR (neat): 2987, 2931, 2857, 1614, 1513, 1463, 1377, 1249, 1087, 1037, 836, 777 cm−1; HRMS (ESI) calcd for [M + Na]+ (C23H38O5SiNa) 445.2386, found 445.2384.
(R)-2-((4S,5S)-2,2-Dimethyl-5-vinyl-1,3-dioxolan-4-yl)-2-((4-methoxybenzyl)oxy)ethanol (6)
Tetrabutylammonium fluoride trihydrate (821 mg, 2.61 mmol) was added to the solution of 5 (1.0 g, 2.37 mmol) and THF (20 mL). The reaction mixture was stirred at rt for 2 h and concentrated. The crude product was further purified by column chromatography (SiO2, EtOAc–hexanes, 1:2; Rf 0.30) to give 6 (599 mg, 1.95 mmol, 82%) as a colourless oil. [α]D20 −17.5 (c 1.2, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 1.35 (s, 3H), 1.46 (s, 3H), 2.08 (br, 1H), 3.46 (m, 1H), 3.78 (s, 3H), 3.80–3.88 (m, 2H), 4.24 (dd, J = 9.0 Hz, J = 8.7 Hz, 1H), 4.36 (d, J = 10.5 Hz, 1H), 4.47 (d, J = 10.5 Hz, 1H), 4.70 (dd, J = 6.4 Hz, J = 6.4 Hz, 1H), 5.25 (dd, J = 10.6 Hz, J = 1.4 Hz, 1H), 5.39 (dd, J = 17.1 Hz, J = 1.4 Hz, 1H), 5.91 (ddd, J = 6.5 Hz, J = 10.5 Hz, J = 17.0 Hz, 1H), 6.85 (d, J = 8.6 Hz, 2H), 7.21 (d, J = 8.6 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 159.4, 133.9, 129.9, 129.4, 117.2, 113.9, 108.6, 78.5, 77.4, 77.1, 71.0, 61.3, 55.3, 27.7, 25.3; IR (neat): 3471, 2987, 2935, 1612, 1513, 1461, 1378, 1249, 1072, 1035, 873, 823, 514 cm−1. HRMS (MALDI) calcd for [M + Na]+ (C17H24O5Na) 331.1522, found 331.1536.
(5R,6R)-8,8,9,9-Tetramethyl-5-((R)-oxiran-2-yl)-6-vinyl-2,4,7-trioxa-8-siladecane (7a)
Dimethyl sulfoxide (126 μL, 1.62 mmol) was added to the solution of oxalyl chloride (67 μL, 0.81 mmol) and dichloromethane (3 mL) at −78 °C. The reaction mixture was stirred at −78 °C for 30 min, then added to a solution of 6 (100 mg, 0.324 mmol) and dichloromethane (0.5 mL), stirred for another 40 min at −78 °C and added with triethylamine (489 μL). The mixture was warmed up to rt, diluted with CH2Cl2 (25 mL) and washed with water (5 mL × 3). The organic layer was separated, dried over Na2SO4, filtered and concentrated. The crude product was further purified by column chromatography (SiO2, EtOAc–hexanes, 1:2; Rf 0.60) to give 7a (80 mg, 0.26 mmol, 80%) as a light yellow oil. [α]D20 13.3 (c 0.87, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 1.33 (s, 3H), 1.47 (s, 3H), 3.74 (dd, J = 7.8 Hz, J = 2.8 Hz, 1H), 3.78 (s, 3H), 4.32–4.37 (m, 2H), 4.45 (d, J = 10.5 Hz, 1H), 4.72 (t, J = 6.2 Hz, J = 6.2 Hz, 1H), 5.25 (dd, J = 10.5 Hz, J = 1.4 Hz, 1H), 5.42 (dd, J = 17.1 Hz, J = 1.4 Hz, 1H), 5.89 (ddd, J = 6.2 Hz, J = 10.6 Hz, J = 16.9 Hz, 1H), 6.85 (d, J = 8.6 Hz, 2H), 7.23 (d, J = 8.4 Hz, 2H), 9.61 (d, J = 2.8 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ 200.9, 159.6, 132.7, 130.1, 128.9, 118.0, 113.9, 109.6, 81.5, 78.3, 77.0, 72.4, 55.3, 27.4, 25.2; IR (neat): 2987, 2935, 2838, 1733, 1610, 1513, 1461, 1378, 1251, 1074, 1035, 871, 825, 514 cm−1. HRMS (MALDI) calcd for [M + Na]+ (C17H22O5Na) 329.1365, found 329.1552.
(R)-2-((tert-Butyldimethylsilyl)oxy)-2-((4S,5S)-2,2-dimethyl-5-vinyl-1,3-dioxolan-4-yl)ethyl pivalate (9)
Tert-butyldimethylsilyl chloride (872 mg, 5.78 mmol) was added to the solution of 824 (1.05 g, 3.85 mmol), imidazole (393 mg, 5.78 mmol), 4-(dimethylamino)pyridine (47 mg, 0.38 mmol) and dichloromethane (25 mL). The reaction mixture was stirred at 40 °C for 14 h and concentrated. The crude product was further purified by column chromatography (SiO2, EtOAc–hexanes, 1:9; Rf 0.70) to give 9 (1.12 g, 2.89 mmol, 75%) as a colourless liquid. [α]D20 −10.75 (c 0.9, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 0.03 (s, 3H), 0.04 (s, 3H), 0.84 (s, 9H), 1.19 (s, 9H), 1.31 (s, 3H), 1.42 (s, 3H), 3.91 (m, 1H), 4.07–4.21 (m, 3H), 4.57 (t, J = 6.9 Hz, J = 6.9 Hz, 1H), 5.22 (d, J = 10.4 Hz, 1H), 5.34 (d, J = 17.1 Hz, 1H), 5.92 (ddd, J = 7.5 Hz, J = 10.2 Hz, J = 17.4 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ 178.3, 134.3, 118.3, 108.4, 78.7, 77.8, 69.4, 66.1, 38.9, 27.8, 27.2, 25.8, 25.3, 18.0, −3.9, −4.6. HRMS (ESI) calcd for [M + Na]+ (C20H38O5SiNa) 409.2386, found 409.2393.
(R)-2-((tert-Butyldimethylsilyl)oxy)-2-((4S,5S)-2,2-dimethyl-5-vinyl-1,3-dioxolan-4-yl)ethanol (10)
Diisobutylaluminium hydride (1.1 M in cyclohexane, 2.93 mL, 3.23 mmol) was added dropwise to the solution of 9 (500 mg, 1.29 mmol) and dichloromethane (20 mL) at −78 °C. The reaction mixture was stirred at −78 °C for another 2 h, warmed up to rt, quenched with methanol (3 mL) and citric acid(aq) (w/w, 10%, 5 mL), concentrated. The residue was added with water (10 mL) and extracted with dichloromethane (12 mL × 3). The organic layers were combined, dried over Na2SO4, filtered and concentrated. The crude product was further purified by column chromatography (SiO2, EtOAc–hexanes, 1:7; Rf 0.33) to give 10 (308 mg, 1.02 mmol, 79%) as a colourless liquid. [α]D20 −24.2 (c 1.1, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 0.04 (s, 3H), 0.07 (s, 3H), 0.85 (s, 9H), 1.34 (s, 3H), 1.44 (s, 3H), 2.16 (br, 1H), 3.68–3.81 (m, 3H), 4.16 (t, J = 13.8 Hz, J = 13.8 Hz, 1H), 4.59 (t, J = 6.6 Hz, J = 6.6 Hz, 1H), 5.22 (d, J = 10.7 Hz, 1H), 5.34 (d, J = 17.1 Hz, 1H), 5.90 (ddd, J = 7.2 Hz, J = 10.2 Hz, J = 17.2 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ 134.1, 118.4, 108.5, 79.5, 78.8, 70.8, 65.0, 27.8, 25.9, 25.4, 18.1, −3.7, −4.4. HRMS (ESI) calcd for [M + Na]+ (C15H30O4SiNa) 325.1811, found 325.1807.
(S)-2-((tert-Butyldimethylsilyl)oxy)-2-((4S,5S)-2,2-dimethyl-5-vinyl-1,3-dioxolan-4-yl)acetaldehyde (7b)
Dimethyl sulfoxide (89 μL, 1.15 mmol) was added to the solution of oxalyl chloride (47 μL, 0.58 mmol) and dichloromethane (3 mL) at −78 °C. The reaction mixture was stirred at −78 °C for 30 min, then added with a solution of 10 (87 mg, 0.29 mmol) and dichloromethane (0.5 mL), stirred for another 40 min at −78 °C and added with triethylamine (400 μL). The mixture was warmed up to rt, diluted with CH2Cl2 (20 mL) and washed with water (5 mL × 3). The organic layer was separated, dried over Na2SO4, filtered and concentrated. The crude product was further purified by column chromatography (SiO2, EtOAc–hexanes, 1:7; Rf 0.65) to give 7b (77 mg, 0.26 mmol, 90%) as a colourless oil. [α]D20 0.5 (c 0.6, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 0.03 (s, 3H), 0.07 (s, 3H), 0.88 (s, 9H), 1.31 (s, 3H), 1.45 (s, 3H), 4.08 (m, 1H), 4.35 (t, J = 6.8 Hz, J = 6.8 Hz, 1H), 4.64 (t, J = 6.8 Hz, J = 6.8 Hz, 1H), 5.23 (dd, J = 10.4 Hz, J = 1.2 Hz, 1H), 5.32 (dd, J = 17.2 Hz, J = 1.2 Hz, 1H), 5.98 (ddd, J = 7.3 Hz, J = 10.2 Hz, J = 17.4 Hz, 1H), 9.57 (s, 1H); 13C NMR (CDCl3, 75 MHz) δ 201.8, 134.0, 119.2, 109.2, 79.3, 78.6, 76.7, 27.1, 25.8, 24.9, 18.1, −4.3, −4.7. HRMS (ESI) calcd for [M + H]+ (C15H29O4Si) 301.1835, found 301.1831.
General procedure for the vinylalumination of aldehydes
Methyl propiolate (204 μL, 3.23 mmol) was added to the solution of diisobutylaluminum hydride (1.1 M in cyclohexane, 2.67 mL, 2.94 mmol), HMPA (766 μL, 4.41 mmol) and THF 10 mL at 0 °C. After being stirred at 0 °C for 1 h, the reaction mixture became a yellow solution and was warmed up to rt in 20 min. A solution of 7a (300 mg, 0.98 mmol) and THF (4 mL) was added to the above solution of [α-(methoxycarbonyl)vinyl]diisobutylaluminum (1a). The resulting mixture was stirred for another 14 h at rt, quenched with methanol (3 mL), added with citric acid(aq) (10%, w/w), stirred for 3 min, concentrated to remove THF and extracted with diethyl ether (10 mL × 3). The organic layers were separated, dried over Na2SO4, filtered and concentrated. The crude product was further purified by column chromatography (SiO2, EtOAc–hexanes, 1:3; 11a, Rf 0.30; 12a, Rf 0.25) to give 11a (184 mg, 0.47 mmol, 49%) and 12a (46.3 mg, 0.12 mmol, 12%) as colourless oils.
11a
.
[α]D20 −26.9 (c 1.02, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 1.29 (s, 3H), 1.43 (s, 3H), 3.67 (s, 3H), 3.77 (s, 3H), 3.78–3.82 (m, 1H), 4.14 (dd, J = 8.0 Hz, J = 6.7 Hz, 1H), 4.42 (d, J = 10.5 Hz, 1H), 4.60–4.64 (m, 2H), 4.78 (dd, J = 5.1 Hz, J = 3.9 Hz, 1H), 5.24 (dd, J = 10.9 Hz, J = 1.2 Hz, 1H), 5.34 (dd, J = 17.2 Hz, J = 1.2 Hz, 1H), 5.90–6.02 (m, 2H), 6.24 (d, J = 0.9 Hz, 1H), 6.83 (d, J = 8.7 Hz, 2H), 7.17 (d, J = 8.7 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 167.6, 159.2, 138.3, 134.7, 130.1, 129.4, 126.3, 117.7, 113.7, 108.6, 79.6, 79.1, 77.4, 72.9, 72.8, 55.3, 51.8, 27.9, 25.3; IR (neat): 3482, 2987, 2937, 1718, 1614, 1513, 1440, 1378, 1249, 1079, 1035, 871, 819, 516 cm−1; HRMS (MALDI) calcd for [M + Na]+ (C21H28O7Na) 415.1733, found 415.1744.
12a
.
[α]D20 −26.51 (c 1.2, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 1.36 (s, 3H), 1.54 (s, 3H), 3.06 (br, 1H, OH), 3.72 (s, 3H), 3.77 (s, 3H), 3.78 (m, 1H), 4.23 (dd, J = 8.7 Hz, J = 6.0 Hz, 1H), 4.32 (d, J = 9.9 Hz, 1H), 4.42 (d, J = 9.9 Hz, 1H), 4.69 (t, J = 6.1 Hz, J = 6.1 Hz, 1H), 4.94 (d, J = 1.3 Hz, 1H), 5.25 (dd, J = 10.6 Hz, J = 1.5 Hz, 1H), 5.40 (dd, J = 17.2 Hz, J = 1.5 Hz, 1H), 5.89–5.98 (m, 1H), 6.00 (t, J = 1.3 Hz, J = 1.3 Hz, 1H), 6.34 (t, J = 1.3 Hz, 1H), 6.82 (d, J = 8.7 Hz, 2H), 7.12 (d, J = 8.7 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 166.7, 159.3, 141.4, 133.8, 129.8, 129.4, 125.6, 117.3, 113.8, 108.9, 78.6, 77.2, 72.8, 72.5, 69.3, 55.3, 51.9, 27.8, 25.5; IR (neat): 3498, 2989, 2950, 1720, 1612, 1513, 1440, 1378, 1249, 1179, 1079, 1035, 871, 821, 514 cm−1; HRMS (ESI) calcd for [M + Na]+ (C21H28O7Na) 415.1733, found 415.1731.
General procedure for the vinylalumination of aldehydes with alkali perchlorates
The premixed solution of 7a (300 mg, 0.98 mmol), lithium perchlorate (417 mg, 3.92 mmol, 4 equiv.) and THF (6 mL) was added to the solution of 1a, prepared from methyl propiolate (272 μL, 4.31 mmol), DIBALH (1.1 M in cyclohexane, 3.56 mL, 3.92 mmol), HMPA (1.02 mL, 5.88 mmol) and THF (12 mL), as describe above. The reaction mixture was stirred at rt for 14 h, and the same work-up procedure was followed to give 11a (77 mg, 0.19 mmol, 20%) and 12a (154 mg, 0.39 mmol, 40%).
11b and 12b.
The procedure used to prepare 11a and 12a was followed. Starting with 7b (294 mg, 0.98 mmol), methyl propiolate (204 μL, 3.23 mmol), HMPA (766 μL, 4.41 mmol) and diisobutylaluminium hydride (1.1 M in cyclohexane, 2.67 mL, 2.94 mmol), the inseparable mixture of 11b and 12b (106 mg, 0.27 mmol, 28%) was produced after column chromatography (SiO2, EtOAc–hexanes, 1:8; Rf 0.45) as a colourless oil. HRMS (ESI) calcd for [M + H]+ (C19H35O6Si) 387.2197, found 387.2200.
(3R,4R)-Methyl 4-((4S,5S)-2,2-dimethyl-5-vinyl-1,3-dioxolan-4-yl)-3-methoxy-4-((4-methoxybenzyl)oxy)-2-methylenebutanoate (13)
A solution of 11a (50 mg, 0.13 mmol), iodomethane (25 μL, 0.38 mmol) and THF (1 mL) was added to the suspension of potassium hydride (15 mg, 0.38 mmol) and THF (2.5 mL) at 0 °C. The reaction mixture was stirred for another 20 min, quenched with sat. NH4Cl(aq) (7 mL), concentrated to remove THF and extracted with ethyl acetate (5 mL × 3). The combined organic layers were dried over Na2SO4, filtered and concentrated. The crude product was further purified by column chromatography (SiO2, EtOAc–hexanes, 1:3; Rf 0.50) to give 13 (32 mg, 0.079 mmol, 62%) as a colourless oil. [α]D20 −16.6 (c 0.7, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 1.30 (s, 3H), 1.43 (s, 3H), 3.32 (s, 3H), 3.67–3.71 (m, 1H), 3.68 (s, 3H), 3.77 (s, 3H), 4.17 (dd, J = 8.6 Hz, J = 6.2 Hz, 1H), 4.36 (d, J = 10.5 Hz, 1H), 4.50 (d, J = 2.4 Hz, 1H), 4.59 (t, J = 6.5 Hz, J = 6.5 Hz, 1H), 4.65 (d, J = 10.5 Hz, 1H), 5.19 (ddd, J = 10.4 Hz, J = 1.8 Hz, J = 1.5 Hz, 1H), 5.33 (ddd, J = 17.2 Hz, J = 1.8 Hz, J = 1.5 Hz, 1H), 5.89–6.01 (m, 2H), 6.36 (d, J = 1.4 Hz, 1H), 6.81 (d, J = 8.7 Hz, 2H), 7.17 (d, J = 8.7 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 167.2, 159.1, 137.4, 134.6, 130.5, 129.4, 126.9, 116.9, 113.6, 108.4, 80.8, 79.4, 78.9, 77.19, 72.7, 57.7, 55.3, 51.7, 27.9, 25.4; IR (neat): 2987, 2935, 1718, 1612, 1513, 1440, 1371, 1247, 1081, 1035, 873, 821, 514 cm−1; HRMS (ESI) calcd for [M + Na]+ (C22H30O7Na) 429.1889, found 429.1888.
(3S,4R)-Methyl 4-((4S,5S)-2,2-dimethyl-5-vinyl-1,3-dioxolan-4-yl)-3-methoxy-4-((4-methoxybenzyl)oxy)-2-methylenebutanoate (14)
The procedure used to prepare 13 was followed. Starting with 12a (50 mg, 0.13 mmol), iodomethane (25 μL, 0.38 mmol) and potassium hydride (15 mg, 0.38 mmol), compound 14 (32 mg, 0.079 mmol, 62%) was produced after column chromatography (SiO2, EtOAc–hexanes, 1:3; Rf 0.40) as a colourless oil. [α]D20 −30.3 (c 0.55, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 1.36 (s, 3H), 1.51 (s, 3H), 3.32 (s, 3H), 3.67–3.71 (m, 1H), 3.75 (s, 3H), 3.78 (s, 3H), 4.27–4.40 (m, 4H), 4.66 (t, J = 6.6 Hz, J = 6.6 Hz, 1H), 5.21 (dd, J = 10.6 Hz, J = 1.5 Hz, 1H), 5.37 (dd, J = 17.1 Hz, J = 1.5 Hz, 1H), 5.96 (ddd, J = 6.6 Hz, J = 10.5 Hz, J = 17.1 Hz, 1H), 6.06 (d, J = 1.1 Hz, 1H), 6.42 (d, J = 1.1 Hz, 1H), 6.78 (d, J = 8.6 Hz, 2H), 7.09 (d, J = 8.6 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 166.8, 159.1, 137.3, 134.3, 130.3, 129.1, 127.2, 117.1, 113.6, 108.4, 79.6, 78.7, 77.6, 77.4, 72.3, 57.9, 55.3, 51.9, 28.1, 25.8; IR (neat): 2987, 2935, 1722, 1612, 1513, 1440, 1378, 1249, 1087, 1035, 873, 821, 514 cm−1; HRMS (FAB) calcd for [M + H]+ (C22H31O7) 407.2070, found 407.2068.
(3aS,6R,7R,7aS)-Methyl 6-methoxy-7-((4-methoxybenzyl)oxy)-2,2-dimethyl-3a,6,7,7a-tetrahydrobenzo[d][1,3]dioxole-5-carboxylate (15)
The second generation Grubbs catalyst (10.4 mg, 0.012 mmol) was added to the solution of 13 (50 mg, 0.12 mmol) and toluene (12.8 mL), and the resulting solution was refluxed for 14 h under an atmosphere of nitrogen and concentrated. The crude product was further purified by column chromatography (SiO2, EtOAc–hexanes, 1:2; Rf 0.35) to give 15 (32.3 mg, 0.088 mmol, 74%) as a light brown oil. [α]D20 −45.5 (c 0.65, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 1.35 (s, 3H), 1.39 (s, 3H), 3.47 (dd, J = 4.4 Hz, J = 2.6 Hz, 1H), 3.67 (s, 3H), 3.77 (s, 3H), 3.78 (s, 3H), 4.34 (d, J = 4.4 Hz, 1H), 4.47–4.56 (m, 2H), 4.69 (d, J = 12.3 Hz, 1H), 4.71 (d, J = 12.3 Hz, 1H), 6.71 (d, J = 3.2 Hz, 1H), 6.86 (d, J = 8.6 Hz, 2H), 7.31 (d, J = 8.6 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 166.6, 159.4, 137.1, 130.2, 129.8, 129.5, 113.9, 111.5, 73.9, 72.9, 72.8, 71.2, 70.6, 61.3, 55.3, 52.2, 27.9, 26.2; IR (neat): 2933, 1720, 1614, 1513, 1440, 1376, 1249, 1052, 825, 511, 412 cm−1; HRMS (ESI) calcd for [M + Na]+ (C20H26O7Na) 401.1576, found 401.1581.
(3aS,6S,7R,7aS)-Methyl 6-methoxy-7-((4-methoxybenzyl)oxy)-2,2-dimethyl-3a,6,7,7a-tetrahydrobenzo[d][1,3]dioxole-5-carboxylate (16)
The procedure used to prepare 15 was followed. Starting with 14 (50 mg, 0.12 mmol) and the second generation Grubbs catalyst (10.4 mg, 0.012 mmol), compound 16 (31.5 mg, 0.088 mmol, 73%) was produced after column chromatography (SiO2, EtOAc–hexanes, 1:2; Rf 0.45) as a light brown oil. [α]D20 44.45 (c 0.8, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 1.36 (s, 3H), 1.40 (s, 3H), 3.44 (s, 3H), 3.74 (m, 1H), 3.76 (s, 3H), 3.78 (s, 3H), 4.26 (d, J = 6.2 Hz, 1H), 4.42 (dd, J = 5.8 Hz, J = 3.1 Hz, 1H), 4.59–4.64 (m, 2H), 4.70 (d, J = 12.0 Hz, 1H), 6.70 (d, J = 3.0 Hz, 1H), 6.85 (d, J = 8.7 Hz, 2H), 7.28 (d, J = 8.7 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 166.7, 159.3, 135.9, 132.4, 130.1, 129.7, 113.8, 110.3, 75.4, 75.2, 73.9, 72.7, 71.8, 59.8, 55.3, 52.0, 27.1, 25.9; IR (neat): 2985, 2933, 1722, 1612, 1513, 1438, 1375, 1249, 1097, 1033, 858, 821, 516 cm−1; HRMS (ESI) calcd for [M + Na]+ (C20H26O7Na) 401.1576, found 401.1568.
Pericosine B (2b)
Trifluoroacetic acid (1.7 mL) was added to the solution of 15 (20 mg, 0.055 mmol) and methanol (0.66 mL) at 0 °C. The reaction mixture was stirred at rt for 5 h and concentrated. The crude product was further purified by column chromatography (SiO2, methanol–ethyl acetate, 1:4; Rf 0.45) to give 2b (8.9 mg, 0.041 mmol, 74%) as a colourless, viscous oil. [α]D20 24.4 (c 0.65, EtOH); 1H NMR (acetone-d6/D2O, 300 MHz) δ 3.56 (s, 3H), 3.76 (s, 3H), 3.83 (dd, J = 2.0 Hz, J = 4.4 Hz, 1H), 3.99 (m, 1H) 4.22 (m, 1H), 6.71 (dd, J = 2.2 Hz, J = 1.3 Hz, 1H); 13C NMR (acetone-d6, 75 MHz) δ 166.6, 141.7, 130.6, 76.8, 72.4, 69.8, 69.1, 61.4, 52.2; IR (neat): 3421, 2940, 1712, 1438, 1259, 1201, 1070 cm−1; HRMS (ESI) calcd for [M + Na]+ (C9H14O6Na) 241.0688, found 241.0683.
Pericosine C (2c)
The procedure used to prepare 2b was followed. Starting with 16 (20 mg, 0.055 mmol) and trifluoroacetic acid (1.7 mL), compound 2c (9.1 mg, 0.042 mmol, 76%) was produced after column chromatography (SiO2, methanol–ethyl acetate, 1:4; Rf 0.45) as a colourless, viscous oil. [α]D20 82.1 (c 0.5, EtOH); 1H NMR (acetone-d6/D2O, 300 MHz) δ 3.45 (s, 3H), 3.72 (s, 3H), 3.88 (m, 1H), 3.93 (dd, J = 4.8 Hz, J = 2.1 Hz, 1H), 4.17 (d, J = 4.8 Hz, 1H), 4.23 (t, J = 3.3 Hz, J = 3.3 Hz, 1H), 6.70 (d, J = 3.8 Hz, 1H); 13C NMR (acetone-d6, 75 MHz) δ 167.8, 140.7, 131.4, 79.0, 73.0, 70.4, 67.4, 59.5, 52.2; IR (neat): 3428, 2921, 1712,1648, 1440, 1272, 1070 cm−1; HRMS (ESI) calcd for [M + Na]+ (C9H14O6Na) 241.0688, found 241.0686.
(3aS,6R,7R,7aS)-Methyl 6-hydroxy-7-((4-methoxybenzyl)oxy)-2,2-dimethyl-3a,6,7,7a-tetrahydrobenzo[d][1,3]dioxole-5-carboxylate (17)
The procedure used to prepare 15 was followed. Starting with 11a (50 mg, 0.13 mmol) and the second generation Grubbs catalyst (11.0 mg, 0.013 mmol), compound 17 (35.6 mg, 0.098 mmol, 77%) was produced after column chromatography (SiO2, EtOAc–hexanes, 1:2; Rf 0.25) as a light brown oil. [α]D20 −38.78 (c 0.75, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 1.35 (s, 3H), 1.40 (s, 3H), 3.42 (d, J = 10.2 Hz, 1H), 3.53 (dd, J = 4.3 Hz, J = 2.0 Hz, 1H), 3.78 (s, 3H), 3.80 (s, 3H), 4.53–4.65 (m, 3H), 4.81 (m, 1H), 4.87 (d, J = 11.9 Hz, 1H), 6.73 (dd, J = 3.1 Hz, J = 1.1 Hz, 1H), 6.86 (d, J = 8.6 Hz, 2H), 7.32 (d, J = 8.6 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 166.1, 159.5, 135.8, 131.8, 129.8, 129.3, 113.9, 111.5, 75.7, 73.3, 71.8, 69.7, 62.1, 55.3, 52.4, 28.1, 26.4; IR (neat): 3318, 2935, 1722, 1612, 1513, 1438, 1375, 1249, 1106, 1051, 827, 518 cm−1; HRMS (MALDI) calcd for [M + Na]+ (C19H24O7Na) 387.1420, found 387.1431.
(3S,4S,5R,6R)-Methyl 3,4,5,6-tetrahydroxycyclohex-1-enecarboxylate (18)
Trifluoroacetic acid (1.7 mL) was added to the solution of 17 (20 mg, 0.055 mmol) and methanol (0.66 mL) at 0 °C. The reaction mixture was stirred at rt for 5 h and concentrated. The crude product was further purified by column chromatography (SiO2, methanol–ethyl acetate, 1:4; Rf 0.35) to give 18 (7.7 mg, 0.038 mmol, 76%) as a colourless oil. 1H NMR (D2O, 300 MHz) δ 3.84 (dd, J = 4.6 Hz, J = 1.8 Hz, 1H), 3.87 (s, 3H), 4.20 (d, J = 3.8 Hz, 1H), 4.49 (m, 1H), 4.62 (m, 1H), 6.92 (t, J = 1.7 Hz, J = 1.7 Hz, 1H); 13C NMR (D2O, 75 MHz) δ 168.0, 141.1, 131.0, 71.5, 67.9, 65.2, 52.7.9b
Acknowledgements
This research was supported by the National Science Council (NSC 101-2113-M-008-002), Taiwan. We are grateful to Ms Ping-Yu Lin at the Institute of Chemistry, Academia Sinica, and Valuable Instrument Center in National Central University for obtaining mass analysis.
References
-
(a) T. Tsuda, T. Yoshida, T. Kawamoto and T. Saegusa, J. Org. Chem., 1987, 52, 1624 CrossRef CAS;
(b) T. Tsuda, T. Yoshida and T. Saegusa, J. Org. Chem., 1988, 53, 1037 CrossRef CAS.
- Reviews for Morita–Baylis–Hillman reaction:
(a) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659 CrossRef CAS PubMed;
(b) C. G. Lima-Junior and M. L. A. A. Vasconcellos, Bioorg. Med. Chem., 2012, 20, 3954 CrossRef CAS PubMed;
(c) D. Basavaiah and G. Veeraraghavaiah, Chem. Soc. Rev., 2012, 41, 68 RSC;
(d) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447 CrossRef CAS PubMed;
(e) D. Basavaiah, K. V. Rao and R. J. Reddy, Chem. Soc. Rev., 2007, 36, 1581 RSC;
(f) D. Basavaiah, A. J. Rao and T. Satyanarayana, Chem. Rev., 2003, 103, 811 CrossRef CAS PubMed , and references cited therein.
-
(a) P. V. Ramachandran, D. Pratihar, G. Garner and B. C. Raju, Tetrahedron Lett., 2005, 46, 2121 CrossRef CAS;
(b) P. V. Ramachandran, G. Garner and D. Pratihar, Org. Lett., 2007, 9, 4753 CrossRef CAS PubMed;
(c) F. A. Davis, H. Qiu, M. Song and N. V. Gaddiraju, J. Org. Chem., 2009, 74, 2798 CrossRef CAS PubMed.
- P. V. Ramachandran, M. T. Rudd, T. E. Burghardt and M. V. R. Reddy, J. Org. Chem., 2003, 68, 9310 CrossRef CAS PubMed.
-
(a) A. Numata, M. Iritani, T. Yamada, K. Minoura, E. Matsumura, T. Yamori and T. Tsuruo, Tetrahedron Lett., 1997, 38, 8215 CrossRef CAS;
(b) T. Yamada, M. Iritani, M. Ohishi, K. Tanaka, K. Minoura, M. Doi and A. Numata, Org. Biomol. Chem., 2007, 5, 3979 RSC.
-
(a) T. Yamada, M. Iritani, M. Doi, K. Minoura, T. Ito and A. Numata, J. Chem. Soc., Perkin Trans. 1, 2001, 3046 RSC;
(b) T. Yamada, M. Iritani, M. Minoura and A. Numata, J. Antibiot., 2002, 55, 147 CrossRef CAS PubMed;
(c) T. Yamada, M. Iritani, K. Minoura, K. Kawai and A. Numata, Org. Biomol. Chem., 2004, 2, 2131 RSC;
(d) T. Yamada, M. Doi, A. Miura, W. Harada, M. Hiramura, K. Minoura, R. Tanaka and A. Numata, J. Antibiot., 2005, 58, 185 CrossRef CAS PubMed;
(e) T. Yamada, K. Minoura, R. Tanaka and A. Numata, J. Antibiot., 2006, 59, 345 CrossRef CAS PubMed.
-
(a) J. Duchek, D. R. Adams and T. Hudlicky, Chem. Rev., 2011, 111, 4223 CrossRef CAS PubMed;
(b) Y. Usami, H. Ichikawa and M. Arimoto, Int. J. Mol. Sci., 2008, 9, 401 CrossRef CAS PubMed.
- Reported syntheses using quinic acid or shikimic acid as the starting material:
(a) Y. Usami, K. Suzuki, K. Mizuki, H. Ichikawa and M. Arimoto, Org. Biomol. Chem., 2009, 7, 315 RSC;
(b) Y. Usami, I. Takaoka, H. Ichikawa, Y. Horibe, S. Tomiyama, M. Ohtsuka, Y. Imanishi and M. Arimoto, J. Org. Chem., 2007, 72, 6127 CrossRef CAS PubMed;
(c) Y. Usami and K. Mizuki, J. Nat. Prod., 2011, 74, 877 CrossRef CAS PubMed;
(d) Y. Usami, M. Ohsugi, K. Mizuki, H. Ichikawa and M. Arimoto, Org. Lett., 2009, 11, 2699 CrossRef CAS PubMed;
(e) Y. Usami, Y. Horibe, I. Takaoka, H. Ichikawa and M. Arimoto, Synlett, 2006, 1598 CrossRef CAS;
(f) Y. Usami and Y. Ueda, Chem. Lett., 2005, 34, 1062 CrossRef CAS;
(g) Y. Usami and Y. Ueda, Synthesis, 2007, 3219 CrossRef CAS;
(h) Y. Usami, K. Mizuki, H. Ichikawa and M. Arimoto, Tetrahedron: Asymmetry, 2008, 19, 1461 CrossRef CAS.
- Using 3,5-cyclohexadiene-cis-1,2-diols:
(a) T. J. Donohoe, K. Blades, M. Helliwell and M. J. Waring, Tetrahedron Lett., 1998, 39, 8755 CrossRef CAS;
(b) D. R. Boyd, N. D. Sharma, C. A. Acaru, J. F. Malone, C. R. O'Dowd, C. C. R. Allen and P. J. Stevenson, Org. Lett., 2010, 12, 2206 CrossRef CAS PubMed.
- Syntheses of related cyclitol/carbasugar natural products:
(a) M. J. Palframan, G. Kociok-Köhn and S. E. Lewis, Chem.–Eur. J., 2012, 18, 4766 CrossRef CAS PubMed;
(b) M. J. Palframan, G. Kociok-Köhn and S. E. Lewis, Org. Lett., 2011, 13, 3150 CrossRef CAS PubMed;
(c) S. Pilgrim, G. Kociok-Köhn, M. D. Lloyd and S. E. Lewis, Chem. Commun., 2011, 47, 4799 RSC;
(d) M. Ali Khan, J. P. Lowe, A. L. Johnson, A. J. W. Stewart and S. E. Lewis, Chem. Commun., 2011, 47, 215 RSC;
(e) D. R. Adams, C. Aichinger, J. Collins, U. Rinner and T. Hudlický, Synlett, 2011, 725 CAS.
- Y. S. Reddy, P. Kadigachalam, R. K. Basak, A. P. J. Pal and Y. D. Vankar, Tetrahedron Lett., 2012, 53, 132 CrossRef CAS.
- S. Tripathi, A. C. Shaikh and C. Chen, Org. Biomol. Chem., 2011, 9, 7306 CAS.
- One report showed the influence of β-substituent: R. B. Andrade and S. F. Martin, Org. Lett., 2005, 7, 5733 CrossRef CAS PubMed.
- H. R. Moon, W. J. Choi, H. O. Kim and L. S. Jeong, Tetrahedron: Asymmetry, 2002, 13, 1189 CrossRef CAS.
- K. P. Jang, S. Y. Choi, Y. K. Chung and E. Lee, Org. Lett., 2011, 13, 2476 CrossRef CAS PubMed.
- A. J. Mancuso and D. Swern, Synthesis, 1981, 165 CrossRef CAS.
- V. A. Stenger, J. Chem. Eng. Data, 1996, 41, 1111 CrossRef CAS.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953 CrossRef CAS PubMed.
-
(a) D. R. Williams, J. L. Moore and M. Yamada, J. Org. Chem., 1986, 51, 3916 CrossRef CAS;
(b) A. Dondoni and P. Merino, J. Org. Chem., 1991, 56, 5294 CrossRef CAS;
(c) S. Chandrasekhar, M. S. Reddy, G. S. Kiranbabu and A. S. K. Murthy, Tetrahedron Lett., 2006, 47, 7307 CrossRef CAS.
-
(a) M. Chérest, H. Felkin and N. Prudent, Tetrahedron Lett., 1968, 9, 2199 CrossRef;
(b) N. T. Anh and O. Eisenstein, Nouv. J. Chim., 1977, 1, 61 Search PubMed;
(c) N. T. Anh, O. Eisenstein, J.-M. Lefour and M.-E. Dau, J. Am. Chem. Soc., 1973, 95, 6146 CrossRef;
(d) N. T. Anh and O. Eisenstein, Tetrahedron Lett., 1976, 155 CrossRef.
- It is interesting to know that the major products derived from the nucleophilic addition of 2-O-benzyl-3,4-O-(pent-3-ylidene)-threose were the syn-adducts. See:
(a) X. Lu and R. Bittman, Tetrahedron Lett., 2005, 46, 3165 CrossRef CAS;
(b) X. Lu, H.-S. Byun and R. Bittman, J. Org. Chem., 2004, 69, 5433 CrossRef CAS PubMed.
- D. J. Cram and D. R. Wilson, J. Am. Chem. Soc., 1963, 85, 1245 CrossRef CAS.
- A. Mengel and O. Reiser, Chem. Rev., 1999, 99, 1191 CrossRef CAS PubMed.
- V. Navickas and M. E. Maier, Tetrahedron, 2010, 66, 94 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of the 1H NMR and 13C NMR spectra for the new compounds. See DOI: 10.1039/c3ra45871g |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.