
Random copolymerisations catalysed by simple titanium α-amino acid complexes†
Abstract
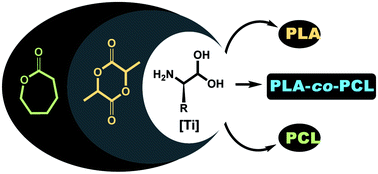
α-Amino acid complexes of titanium have been applied in the synthesis of biopolymers. The complexes can be prepared under mild conditions often using hydrous procedures and result in the preparation of polylactide and polycaprolactone without the need for rigorous drying and air-sensitive handling. The complexes also prove to be excellent initiators for the synthesis of random copolymers: they are rare examples of completely air stable designed catalysts to undertake such a transformation. The composition and thermal properties of the random copolymers are also investigated.
 Please wait while we load your content...
Please wait while we load your content...

