Ultrasound-assisted C–C coupling reactions catalyzed by unique SPION-A-Pd(EDTA) as a robust nanocatalyst
Marzieh Ghotbinejad,
Ahmad R. Khosropour*,
Iraj Mohammadpoor-Baltork*,
Majid Moghadam,
Shahram Tangestaninejad and
Valiollah Mirkhani
Catalysis Division, Department of Chemistry, University of Isfahan, 81746-73441, Isfahan, Iran. E-mail: khosropour@chem.ui.ac.ir; imbaltork@sci.ui.ac.ir; Fax: +98 3116689732; Tel: +98 3117932700
First published on 5th December 2013
Abstract
A novel and highly stable Pd(EDTA)2− salt was synthesized as a catalyst, using a counter-cation of N-methylimidazolium bonded to 1,3,5-triazine-tethered SPIONs (superparamagnetic iron oxide nanoparticles). This well-defined complex efficiently catalyzed the Mizoroki–Heck and Suzuki–Miyaura cross-coupling reactions. The cross-coupled products were produced under conventional heating and ultrasound irradiation at an extremely low catalyst loading (as low as 0.032 mol% Pd). Results indicated that conventional synthesis took longer and gave moderate yields, while in the presence of ultrasound irradiation, the reaction occurred very fast in high to excellent yields. The catalyst could be quickly recovered by an external magnetic field and could be reused for several reaction cycles without any change in catalytic activity.
Introduction
With the increasing environmental consciousness in chemical research, the challenge of designing sustainable environmentally friendly procedures has become the fundamental aim of green chemistry. From the first report on using ultrasound in organic synthesis,1 the motivation to use this technique in organic transformations increased tremendously. Ultrasonic-assisted organic synthesis (UAOS) is a promisingly useful approach to a green technique in organic synthesis.2–4 The physical and chemical effects of ultrasound irradiation are based on acoustic cavitations resulting from the continuous formation, growth and implosive collapse of bubbles in a solution, which results in a high temperature and pressure pulse.5–8 The advantages of ultrasound irradiation in organic synthesis are the formation of purer products in good yields, short reaction times, improved energy conservation, easier manipulation, mild conditions and waste minimization which is comparable to traditional methods.9–15The palladium catalyzed cross-coupling reactions are the most powerful and selective tools for carbon–carbon bond formation.16–18 These cross-coupling reactions have been abundantly used in organic synthesis, pharmaceuticals, and the synthetic methodologies of natural products, conducting polymers, and liquid crystals.19,20 Among these reactions, the Mizoroki–Heck21,22 and Suzuki–Miyaura23 reactions have been recognized as important tools in modern organic synthesis.
The Heck reaction is a transformation between aryl halides and olefins that leads to the formation of disubstituted olefins.24,25 The Suzuki reaction is the most powerful method for coupling aryl halides with phenylboronic acid, which provides an effective method for synthesizing biaryls.26–30
These reactions generally proceed in the presence of a homogeneous palladium catalyst. The difficulties in product separation and recycling of the catalyst have limited the applications of homogeneous palladium catalysts in recent years.31,32 Therefore, in order to overcome these drawbacks, many investigations on developing effective methods for immobilization of Pd complexes on different solid supports, such as microporous polymers,33 activated carbon,34 clays,35 and magnetic nanoparticles (MNPs) have been performed. Magnetic nanoparticle-supported catalysts are the better choice as they not only show excellent catalytic activities but the magnetic nature of these particles also allows for facile recovery and recycling of the catalyst without use of the traditional filtration method.36,37 Moreover, surface modified superparamagnetic iron oxide nanoparticles (SPIONs) have received increasing interest in the past few years both in biomedical and organic transformations.38 Very recently, we reported synthesis of SPION-ACl2 as a green and powerful nano-catalyst for the efficient synthesis of Betti bases.39 It was found that due to the high magnetization of the catalyst it could be satisfactory recovered by a simple external magnet. Moreover, the catalyst could be easily recycled and reused without a loss of its activity.
Now, encouraged by the previous results and our interest in developing efficient, sustainable and greener pathways for organic transformations,40 particularly those under ultrasonic irradiation41 and C–C coupling reactions,42 we would like to report herein a new and powerful palladium-EDTA complex-tagged dicationic ionic liquid with a 1,3,5-triazine core anchored to superparamagnetic nanoparticles (SPION-ACl2), and its application in cross-coupling reactions such as the Mizoroki–Heck and Suzuki–Miyaura reactions, under ultrasound irradiation.
Result and discussion
The synthetic pathway for the catalyst is depicted in Scheme 1.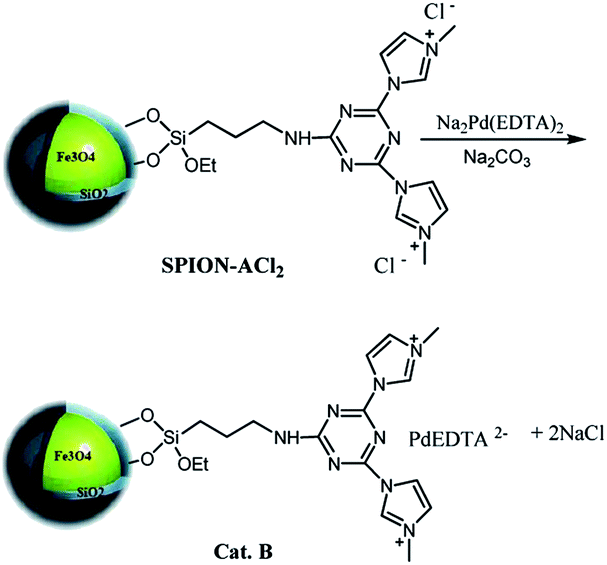 | ||
| Scheme 1 The synthetic pathway for SPION-A-Pd(EDTA). | ||
SPION-A-Pd(EDTA) was characterized by means of Fourier transform infrared spectroscopy (FT-IR), inductively coupled plasma atomic emission spectroscopy (ICP) thermal gravimetric analysis (TGA), and high resolution transmission electron microscopy (HR-TEM).
Fig. 1 illustrates the FT-IR spectra of Fe3O4 (a), silica-encapsulated Fe3O4 (b), and the nanocatalyst SPION-A-Pd(EDTA) (c), respectively.
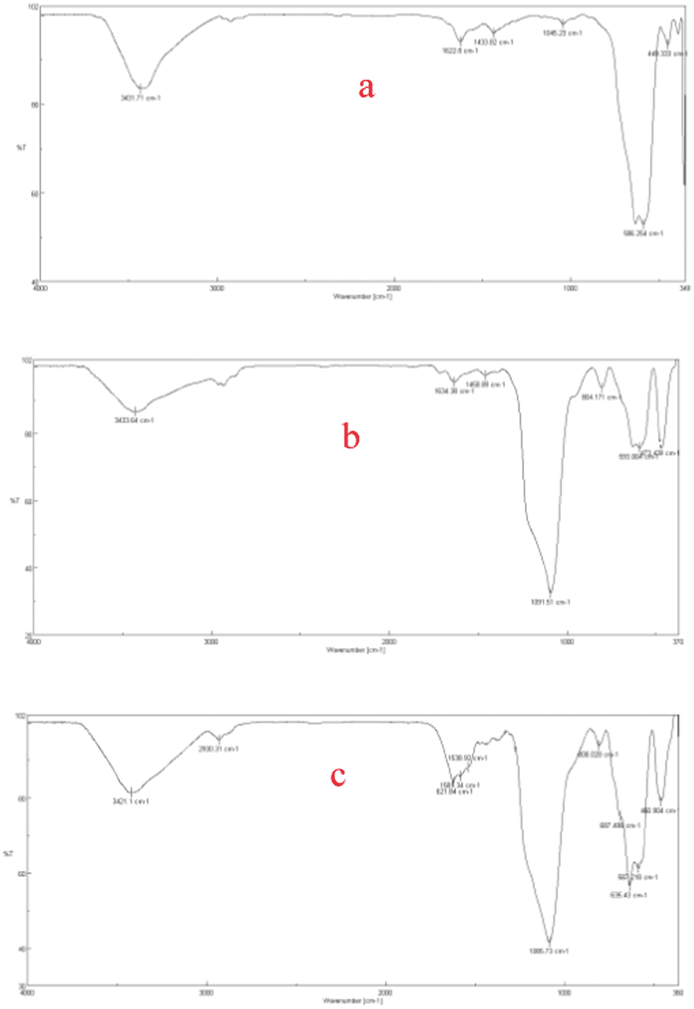 | ||
| Fig. 1 Comparison of the FT-IR spectra of (a) Fe3O4; (b) silica-encapsulated Fe3O4; (c) SPION-A-Pd(EDTA). | ||
The FT-IR spectrum of SPION-A-Pd(EDTA) (Fig. 1c) showed absorption bands at 3421 cm−1 (N–H stretching vibration), 2930 cm−1 (C–H), 1622 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and 635–587 cm−1 (Fe–O) SPIONs.
N) and 635–587 cm−1 (Fe–O) SPIONs.
Inductively coupled plasma atomic emission spectroscopy (ICP) determined the amount of palladium in SPION-A-Pd(EDTA) as 3.41 wt%.
The thermal stability of SPION-A-Pd(EDTA) was also evaluated by thermal gravimetric analysis–differential thermal analysis (TGA-DTG). According to this curve, two weight loss steps were observed. In the first step (below 180 °C), the water molecules (4.59%) in the structure were omitted, while the organic part (11.28%) was lost between 180 and 480 °C (Fig. 2).
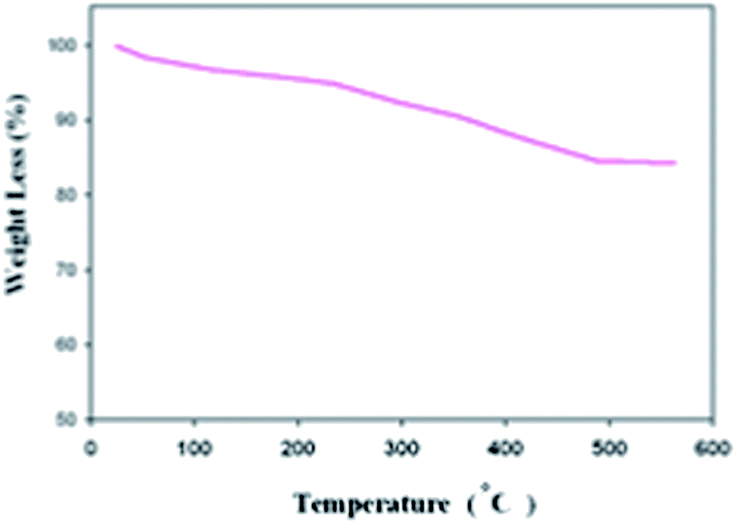 | ||
| Fig. 2 TG-DTG analysis of SPION-A-Pd(EDTA). | ||
To study the morphology of SPION-A-Pd(EDTA), an HR-TEM image was also investigated (Fig. 3).
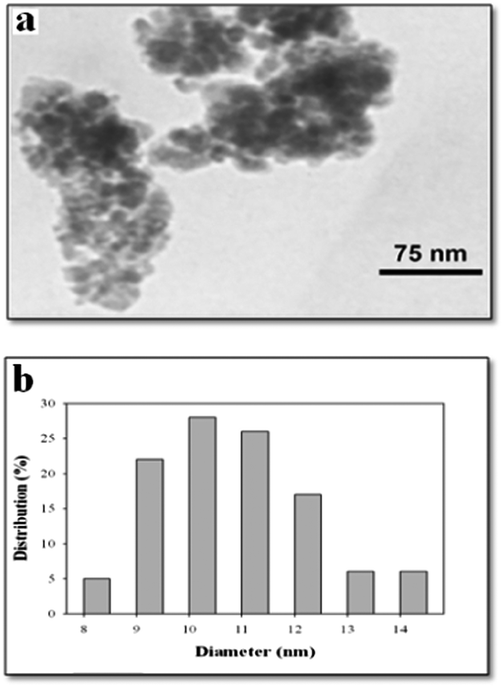 | ||
| Fig. 3 (a) HR-TEM image of SPION-A-Pd(EDTA) and (b) SPION-A-Pd(EDTA) particle size distribution histogram. | ||
HR-TEM images of SPION-A-Pd(EDTA) revealed that it appears to have an almost spherical structure with an average size of about 10–13 nm (Fig. 3b). Thus, the enormous active sites of this nanoparticle may display excellent activity levels in organic transformations.
After the structure characterization of the catalyst, in order to evaluate the catalytic activity of SPION-A-Pd(EDTA), we initially applied it in the Mizoroki–Heck reaction under ultrasound irradiation. To optimization the reaction conditions the reaction of iodobenzene (1.0 mmol) and styrene (1.0 mmol) was chosen as a model, and the role of various bases, solvents and the output power of the ultrasound apparatus were investigated. (Table 1, entries 1–14).
|
||
|---|---|---|
| Entry | Yieldb (%) | |
| a Reaction conditions: Iodobenzene (1 mmol), styrene (1.5 mmol) and K2CO3 (1.5 mmol) in the presence of the catalyst containing 0.003 mol% Pd in 2 mL of DMF.b Isolated yield. | ||
| Solvent effect | ||
| 1 | DMF | 96 |
| 2 | Toluene | 60 |
| 3 | Ethanol | 55 |
| Base effect | ||
| 4 | K2CO3 | 96 |
| 5 | Na2CO3 | 87 |
| 6 | NEt3 | 50 |
| Catalyst effect | ||
| 7 | — | — |
| 8 | Pd(OAc)2@nanoFe3O4 nanoSiO2 | 20 |
| 9 | Pd(OAc)2@nano-SiO2 | 15 |
| 10 | SPION-A-Pd(EDTA) | 96 |
| Power effect (watt) | ||
| 11 | 120 | 78 |
| 12 | 140 | 85 |
| 13 | 170 | 96 |
| 14 | 200 | 96 |
As illustrated in Table 1, DMF was the best solvent for this synthesis. Other solvents such as ethanol and toluene gave only moderate yields of the product (Table 1, entries 1–3).
Among the various bases screened, K2CO3 was found to be the most effective base for this transformation (Table 1, entry 4).
Other bases such as Na2CO3 and NEt3 gave moderate yields of the product (Table 1, entries 5, 6).
No reaction occurred without the catalyst (Table 1, entry 7). More examination revealed that the yield reduced drastically with the replacement of Pd(OAc)2@nano-Fe3O4 or Pd(OAc)2@nano-SiO2 instead of SPION-A-Pd(EDTA) as the catalyst (Table 1, entries 8 and 9).
We also found that the output power of the ultrasound apparatus greatly affected this transformation. The obvious improvement in the conversion (96%) reached a plateau at 170 W of power. Higher acoustic power (200–400 W) made no obvious difference in the yield of the product (Table 1, entry 14) but using lower power (140 W) sharply decreased the conversion to approximately 85% even with more reaction time (Table 1, entry 12).
Accordingly, performing the reaction at 170 W in the presence of 0.094 g of SPION-A-Pd(EDTA) (0.003 mol % Pd), with K2CO3 as the base, and DMF as the solvent, at 50 °C, was optimal for the Mizoroki–Heck reaction. This optimized sonochemical reaction was applied to the synthesis of a variety of disubstituted olefins.
To assess the influence of ultrasonic irradiation on this transformation, we initially examined this reaction under thermal conditions (Table 2). As shown in Table 2, the reaction takes place efficiently in high TOF between 2 and 14 hours at 90 °C.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | X | Product | Time (h) | Yieldb (%) | TOFc |
| a Reactions were carried out under aerobic conditions in 2 ml of mixture of DMF, 1 mmol aryl halide, 1.5 mmol styrene derivative, 1.5 mmol K2CO3 in the presence of SPION-A-Pd(EDTA) (0.003 mol% Pd) and 90 °C.b Isolated yield.c [mol product/mol palladium] h−1. | |||||||
| 1 | H | H | I | 2a | 2 | 87 | 1.4 × 104 |
| 2 | H | 4-Me | I | 2b | 8 | 90 | 3.7 × 103 |
| 3 | H | H | Br | 2a | 7 | 83 | 3.9 × 103 |
| 4 | H | 4-Me | Br | 2b | 9 | 85 | 3.1 × 103 |
| 5 | 4-MeO | 4-Me | I | 2c | 6 | 84 | 4.7 × 103 |
| 6 | 4-Me | H | Br | 2b | 8 | 80 | 3.3 × 103 |
| 7 | 4-Me | 4-Me | Br | 2d | 9 | 83 | 3.1 × 103 |
| 8 | 4-Me | H | I | 2b | 8 | 85 | 3.5 × 103 |
| 9 | 4-Me | 4-Me | I | 2d | 10 | 87 | 2.9 × 103 |
| 10 | 4-Ac | H | I | 2e | 3 | 88 | 9.8 × 103 |
| 11 | 4-Ac | 4-Me | Br | 2f | 5 | 86 | 5.7 × 103 |
| 12 | 4-Ac | H | Br | 2e | 7 | 84 | 4.0 × 103 |
| 13 | 4-Ac | 4-Me | Br | 2f | 8 | 84 | 3.5 × 103 |
| 14 | 4-CHO | H | Br | 2g | 10 | 86 | 2.9 × 103 |
| 15 | 4 F | H | Br | 2h | 14 | 82 | 1.9 × 103 |
| 16 | 4 F | 4-Me | Br | 2i | 12 | 87 | 2.4 × 103 |
To demonstrate the effect of sonication, the synthesis of all the corresponding products was also investigated with ultrasonic irradiation (Table 3). It is apparent that the ultrasound accelerates this transformation under milder conditions.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | X | Product | Time (min) | Yieldb (%) | TOFc |
| a Reactions were carried out under aerobic conditions in 2 ml of mixture of DMF, 1 mmol aryl halide, 1.5 mmol styrene derivative, 1.5 mmol K2CO3 in the presence of SPION-A-Pd(EDTA) (0.003 mol% Pd) and power: 170 W at 50 °C.b Isolated yield.c [Mol product/mol palladium] h−1. | |||||||
| 1 | H | H | I | 2a | 10 | 96 | 1.9 × 105 |
| 2 | H | 4-Me | I | 2b | 8 | 97 | 2.5 × 105 |
| 3 | H | H | Br | 2a | 17 | 88 | 1.0 × 105 |
| 4 | H | 4-Me | Br | 2b | 15 | 91 | 1.2 × 105 |
| 5 | 4-MeO | 4-Me | I | 2c | 20 | 91 | 9.2 × 104 |
| 6 | 4-Me | H | Br | 2b | 19 | 87 | 9.1 × 104 |
| 7 | 4-Me | 4-Me | Br | 2d | 18 | 89 | 9.9 × 104 |
| 8 | 4-Me | H | I | 2b | 16 | 89 | 1.1 × 105 |
| 9 | 4-Me | 4-Me | I | 2d | 15 | 91 | 1.2 × 105 |
| 10 | 4-Ac | H | I | 2e | 25 | 90 | 7.1 × 104 |
| 11 | 4-Ac | 4-Me | Br | 2f | 22 | 90 | 8.1 × 104 |
| 12 | 4-Ac | H | Br | 2e | 26 | 88 | 6.8 × 104 |
| 13 | 4-Ac | 4-Me | Br | 2f | 20 | 90 | 9.1 × 104 |
| 14 | 4-CHO | H | Br | 2g | 25 | 88 | 6.9 × 104 |
| 15 | 4 F | H | Br | 2h | 29 | 86 | 5.9 × 104 |
| 16 | 4 F | 4-Me | Br | 2i | 35 | 89 | 5.1 × 104 |
For instance, at the optimal conditions, 1,2-diphenylethylene was produced almost quantitatively (96% isolated yield) with high TOF (5.6 × 105 h-1) after 10 min sonication at 50 °C (Table 3, entry 1), while in silent conditions, a higher temperature (90 °C) was required to obtain the product in only 87% yield and with lower TOF after 2 h (Table 2, entry 1). This achievement could be extended to the other products.
We assume that the beneficial effect of ultrasound on this heterogeneous reaction may be attributed to a better mass transfer and dispersion of the nanocatalyst in the medium in comparison with magnetically stirred reactions, which makes the catalyst more effective in this transformation.
To continue, for the investigation of the effect of the catalyst–sonication combination on the other C–C coupling reactions, we decided to study the Suzuki–Miyaura cross-coupling reaction of arylboronic acids with a variety of aryl halides in the presence of SPION-A-Pd(EDTA) under ultrasonic irradiation.
As shown in Table 4, the optimal conditions included the reaction of iodobenzene (1 mmol), phenylboronic acid (1.1 mmol), K2CO3 (1.5 mmol) and SPION-A-Pd(EDTA) (0.094 g, 0.003 mol% Pd) in DMF/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) under ultrasound irradiation at 30 °C.
:2) under ultrasound irradiation at 30 °C.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | Pd (mol%) | Power | Yieldb (%) |
| a H2O/DMF (V/V) 2:1.b Isolated yield. |
|||||
| 1 | DMF | K2CO3 | 0.003 | 170 | 57 |
| 2 | H2O | K2CO3 | 0.003 | 170 | 54 |
| 3 | H2O/DMFa | K2CO3 | 0.003 | 160 | 95 |
| 4 | H2O/DMFa | K2CO3 | 0.003 | 160 | 95 |
| 5 | H2O/DMFa | Na2CO3 | 0.003 | 160 | 65 |
| 6 | H2O/DMFa | NEt3 | 0.003 | 160 | 47 |
| 7 | H2O/DMFa | K2CO3 | 0.003 | 140 | 80 |
| 8 | H2O/DMFa | K2CO3 | 0.003 | 160 | 95 |
| 9 | H2O/DMFa | K2CO3 | 0.003 | 200 | 95 |
As with the aforementioned results, the generality of the reaction condition was also examined. The results revealed that the yields of the corresponding products were comparable under silent conditions and ultrasound irradiation, while the sonication was performed with a swift reaction under milder conditions in comparison to conventional heating (Table 5). The successful production of the biaryl derivatives indicated that this is a powerful procedure for the Suzuki–Miyaura reaction.
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | X | Product | Silent conditionsb | Ultrasonic conditionsc | ||||
| Time (h) | Yieldd (%) | TOFe | Time (min) | Yieldb (%) | TOFe | |||||
| a Reactions were carried out under aerobic conditions in 2 ml of mixture of DMF and water (1:2), 1 mmol aryl halide, 1.1 mmol arylboronic acid and 1.5 mmol K2CO3(1.5 mmol) with SPION-A-Pd(EDTA) (0.003 mol% Pd).b At 70 °C.c Applied power: 160 W at 30 °C.d Isolated yield.e [Mol product per mol palladium] h−1. |
||||||||||
| 1 | H | H | I | 3a | 4 | 92 | 7.7 × 103 | 10 | 95 | 1.9 × 105 |
| 2 | H | 4-MeO | I | 3b | 2 | 93 | 1 × 5 104 | 7 | 96 | 2.7 × 105 |
| 3 | H | H | Br | 3a | 7 | 85 | 4.0 × 103 | 16 | 88 | 1.1 × 105 |
| 4 | H | 4-MeO | Br | 3b | 6 | 87 | 4.8 × 103 | 14 | 90 | 1.3 × 105 |
| 5 | 4-MeO | H | Br | 3b | 8 | 86 | 3.6 × 103 | 19 | 89 | 9.3 × 104 |
| 6 | 4-MeO | 4-MeO | Br | 3c | 7 | 88 | 4.1 × 103 | 15 | 90 | 1.2 × 105 |
| 7 | 4-Me | H | Br | 3d | 8 | 86 | 3.6 × 103 | 16 | 90 | 1.1 × 105 |
| 8 | 4-Me | 4-MeO | Br | 3e | 6 | 89 | 4.9 × 103 | 13 | 92 | 1.4 × 105 |
| 9 | 4 F | H | Br | 3f | 9 | 82 | 3.0 × 103 | 35 | 87 | 5.0 × 104 |
| 10 | 4-Ac | H | I | 3g | 6 | 83 | 4.6 × 103 | 25 | 88 | 6.9 × 104 |
| 11 | 4-Ac | 4-MeO | I | 3h | 5 | 88 | 5.9 × 103 | 20 | 91 | 9.2 × 104 |
| 12 | 4-Me | H | I | 3d | 6 | 88 | 4.9 × 103 | 14 | 90 | 1.3 × 105 |
| 13 | 4-Me | 4-MeO | I | 3e | 5 | 90 | 6.0 × 103 | 13 | 93 | 1.4 × 105 |
| 14 | 4-MeO | H | I | 3b | 3 | 89 | 9.9 × 103 | 17 | 91 | 1.1 × 105 |
| 15 | 4-MeO | 4-MeO | I | 3c | 4 | 90 | 7.5 × 103 | 14 | 92 | 1.3 × 105 |
| 16 | 4-Ac | H | Br | 3g | 8 | 86 | 3.6 × 103 | 23 | 89 | 7.8 × 104 |
| 17 | 4-Ac | 4-MeO | Br | 3h | 11 | 89 | 2.7 × 103 | 18 | 90 | 1.0 × 105 |
| 18 | 4-CHO | H | Br | 3i | 14 | 85 | 2.0 × 103 | 24 | 88 | 7.3 × 104 |
The recovered SPION-A-Pd(EDTA) could also be reused easily using an applied magnetic field after the end of the reaction, without any significant loss of its high catalytic performance. The examination of the ultrasonic-assisted Mizoroki–Heck and Suzuki–Miyaura C–C coupling reactions using SPION-A-Pd(EDTA) was repeated six times to evaluate the catalyst's recyclability and stability (Fig. 4). The results illustrated the excellent stability of the catalyst under the reaction conditions.
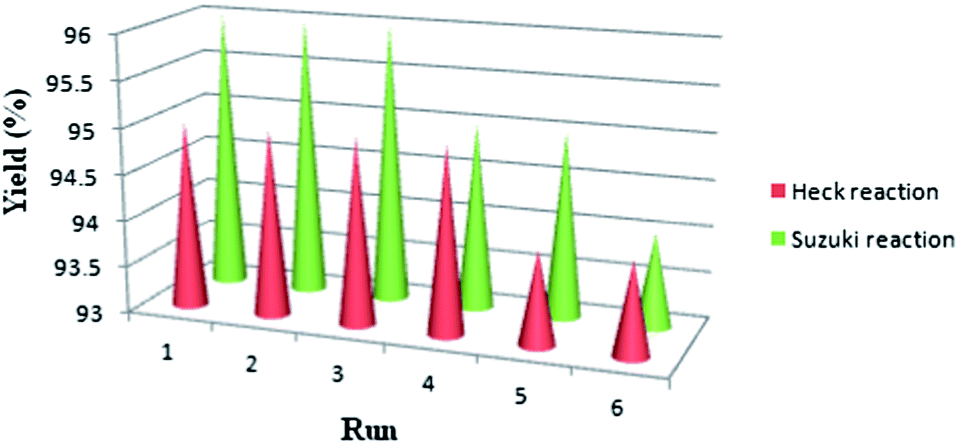 | ||
| Fig. 4 Reuse of SPION-A-Pd(EDTA) examined on the model reactions of Mizoroki–Heck and Suzuki–Miyaura under ultrasonic irradiation. | ||
Initially, the Suzuki–Miyaura cross-coupling of iodobenzene with phenylboronic acid in the presence of SPION-A-Pd(EDTA) was chosen as a template reaction for the optimization of parameters such as base type, solvent and ultrasonic irradiation power of the reaction. The results are summarized in Table 4.
Finally, a comparison of this protocol with recent reports was performed with the Suzuki–Miyaura cross-coupling reaction of phenyl bromide and phenylboronic acid as a template. As shown in Table 6, the best TOF was obtained when utilizing SPION-A-Pd(EDTA).
| Entry | Catalysta | Time (min) | Yieldb (%) | Pd amount (mol %) | TOFc | Ref. |
|---|---|---|---|---|---|---|
| a Reactions were carried out under aerobic conditions in 2 ml of mixture of DMF and water (1:2), 1 mmol phenyl bromide, 1.1 mmol phenylboronic acid and 1.5 mmol K2CO3.b Isolated yield.c [Mol product per mol palladium] h−1. |
||||||
| 1 | SPION-A-Pd(EDTA) | 16 | 88 | 0.003 | 1.1 × 105 | This work |
| 2 | Pd/IL-NH2/SiO2/Fe3O4 | 300 | 87 | 0.5 | 34.8 | 45 |
| 3 | SiO2/BisILsR[PdEDTA] | 600 | 99 | 1 | 99.0 | 44 |
| 4 | Pd–NHC@Fe3O4-IL (3) | 360 | 85 | 0.5 | 28.33 | 46 |
| 5 | Pd(OAc)2@Fe3O4-IL (3) | 360 | 91 | 0.5 | 30.33 | 47 |
| 6 | Fe3O4@SiO2@mSiO2–Pd(0) | 360 | 97 | 0.075 | 215.56 | 48 |
| 7 | SMNPs-Salen Pd(II) | 180 | 100 | 0.5 | 6.67 | 49 |
Experimental
All chemicals were purchased from the Merck chemical company. Fe3O4 nanocomposites and silica-coated magnetite nanoparticles (SiO2@Fe3O4) were synthesised according to the literature.43 The Na2Pd(EDTA) complex was prepared by the dissolution of Pd(OAc)2 (Aldrich), Na2CO3 and Na2H2EDTA (Merck) performed on UV-active aluminum-backed plates of silica gel (TLC Silica gel 60 F254). 1H, and 13C NMR spectra were measured on a Bruker DPX 400 MHz spectrometer in CDCl3 with the chemical shift (δ) given in ppm. Coupling constants are given in Hz.The FT-IR spectra were taken on a Nicolet-Impact 400D spectrophotometer with KBr pellets, and were reported in cm−1. Melting points were determined using a Stuart Scientific SMP2 apparatus and are uncorrected. The sonication was performed in a UP 400S ultrasonic processor equipped with a 3 mm wide and 140 mm long probe, which was immersed directly into the reaction mixture. The operating frequency was 24 kHz and the output power was 0–400 Watts through manual adjustment. The HRTEM images were taken with a Philips CM30 unit operated at 150 kV. The TGA curve was obtained with a heating rate of 10 °C min−1 on a TG 50 Mettler thermogravimetric analyzer from 30 °C to 600 °C. The Pd content of the catalyst was determined by Jarrell-Ash 1100 ICP analysis.
Preparation of SPION-A-Pd(EDTA)
Na2CO3 (0.2 mmol, 0.021 g) was added to a mixture of Na2EDTA (0.1 mmol, 0.037 g) and PdCl2 (0.1 mmol, 0.018 g) in water (5 ml) at 25 °C, and was stirred magnetically for 5 h. In an argon atmosphere, SPION-ACl2 (0.53 g) in EtOH (5 ml) was added dropwise to the solution and the resulting mixture was stirred for a further 12 h at room temperature. Finally, the catalyst was collected by an external permanent magnet, washed with CH2Cl2 (3 × 10 ml) and H2O, and dried under vacuum.General procedure for heterogeneous Heck reactions
A mixture of an aryl halide (1.0 mmol), an alkene (1.5 mmol), K2CO3 (207 mg, 1.5 mmol) and SPION-A-Pd(EDTA) (0.094 g, 0.003 mol % of Pd) in DMF (2 ml) was exposed to ultrasonic irradiation at 170 W at 50 °C for 8–35 min according to Table 3. The progress of the reaction was monitored by TLC (eluent: petroleum ether/ethyl acetate, 4:1). After the completion of the reaction, the catalyst was collected by an external permanent magnet, washed two times with absolute ethanol (2 × 1 ml), air-dried, and used directly for the next round of reactions without further purification. After separation of the catalyst, the collected solution was added to the residue of the reaction mixture and the volatile product was removed in vacuum. The organic residue was washed with water (3 × 10 ml) and dried over anhydrous MgSO4. Purification by flash column chromatography (silica gel, ethyl acetate/petroleum ether) afforded the corresponding products in 86–96% yields.
All products are known in the literature and were identified by comparison of their FT-IR and NMR with literature data. As a sample, the characterization data for 2c is given below.
General procedure for heterogeneous Suzuki reactions
A mixture of an aryl halide (1.0 mmol), an arylboronic acid (1.1 mmol), K2CO3 (207 mg, 1.5 mmol) and Cat. B (0.094 g, 0.003 mol % of Pd) in 2 ml DMF–H2O (1:2 v/v) was exposed to ultrasonic irradiation at 160 W at 30 °C for 7–35 min according to Table 5. The progress of the reaction was monitored by TLC (eluent: petroleum ether–ethyl acetate, 4:1). After the completion of the reaction, the catalyst was collected by an external permanent magnet, washed two times with absolute ethanol (2 × 1 ml), air-dried, and used directly for the next round of the reaction without further purification. After separation of the catalyst, the collected solution was added to the residue of the reaction mixture and the volatile product was removed in vacuum. The organic residue was washed with water (3 × 10 ml) and dried over anhydrous MgSO4. Purification by flash column chromatography (silica gel, ethyl acetate/petroleum ether) afforded the corresponding products in 87–96% yields.
All products are known in the literature and were identified by comparison of their FT-IR, 1H, and 13C NMR with literature data. As a sample, the characterization data for 3c is given below.
Acknowledgements
The support of this work by the Center of Excellence of Chemistry, of the University of Isfahan, (CECUI) is acknowledged.Notes and references
- W. T. Richards and A. L. Loomis, J. Am. Chem. Soc., 1927, 49, 3086 CrossRef CAS.
- G. Cravotto and P. Cintas, Chem. Soc. Rev., 2006, 35, 180 RSC.
- P. A. Vivekand and T. Balakrishnan, Catal. Lett., 2009, 13, 587 CrossRef.
- M. L. Wang and C. J. Chen, Org. Process Res. Dev., 2010, 14, 737 CrossRef CAS.
- P. R. Gogate, Chem. Eng. Proc., 2008, 47, 515 CrossRef CAS.
- V. S. Moholkar, S. P. Sable and A. B. Pandit, AIChE J., 2000, 46, 684 CrossRef CAS.
- W. H. Zhang, X. Quan, J. X. Wang, Z. Y. Zhang and S. Chen, Chemosphere, 2006, 65, 58 CrossRef CAS PubMed.
- K. S. Suslick, M. Fang and T. Hyeon, J. Am. Chem. Soc., 1996, 118, 11960 CrossRef CAS.
- V. V. Namboodiri and R. S. Varma, Org. Lett., 2002, 4, 3161 CrossRef CAS PubMed.
- K. S. Suslick, Science, 1990, 249, 1439 Search PubMed.
- T. J. Mason and J. P. Lorimer, Sonochemistry, John Wiley and Sons, New York, 1988 Search PubMed.
- A. Weissler, H. W. Coofer and S. Synder, J. Am. Chem. Soc., 1950, 72, 1976 Search PubMed.
- J. L. Luche, Synthetic Organic Sonochemistry, Plenum Press, New York, 1998, 10 Search PubMed.
- D. Nagaraja and M. A. Pasha, Tetrahedron Lett., 1999, 40, 7855 CrossRef CAS.
- M. A. Pasha and V. P. Jayashankara, Ultrason. Sonochem., 2006, 13, 42 CrossRef CAS PubMed.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) R. B. Bedford, C. S. J. Cazin and D. Holder, Coord. Chem. Rev., 2004, 248, 2283 CrossRef CAS; (c) A. E. Wang, J. H. Xie, L. X. Wang and Q. L. Zhou, Tetrahedron, 2005, 61, 259 CrossRef CAS; (d) A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020 CrossRef CAS; (e) V. P. W. Bohm, C. W. K. Gstottmayr, T. Weskamp and W. A. Herrmann, J. Organomet. Chem., 2000, 595, 186 CrossRef CAS; (f) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS PubMed.
- (a) V. Farina, Adv. Synth. Catal., 2004, 346, 1553 CrossRef CAS; (b) A. R. Muci and S. L. Buchwald, Top. Curr. Chem., 2002, 219, 131 CrossRef CAS; (c) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed. Engl., 2002, 41, 4176 CrossRef CAS; (d) C. Ballie and J. Xiao, Tetrahedron, 2004, 60, 4159 CrossRef; (e) N. J. Whitcombe, K. K. Hii and S. E. Gibson, Tetrahedron, 2001, 57, 7449 CrossRef CAS.
- (a) A. Häberli and C. J. Leumann, Org. Lett., 2001, 3, 489 CrossRef; (b) P. Lloyd-Williams and E. Giralt, Chem. Soc. Rev., 2001, 30, 145 RSC; (c) R. B. Bedford, Chem. Commun., 2003, 15, 1787 RSC; (d) C. Najera, J. Gil-Molto, S. Karlstrom and L. R. Falvello, Org. Lett., 2003, 5, 1451 CrossRef CAS PubMed; (e) N. T. S. Phan, M. V. D. Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609 CrossRef CAS.
- (a) V. Resch, J. H. Schrittwieser, E. Siirola and W. Kroutil, Curr. Opin. Biotech., 2011, 22, 1 CrossRef PubMed; (b) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS PubMed; (c) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed; (d) R. E. Minto and B. J. Blacklock, Prog. Lipid. Res., 2008, 47, 233 CrossRef CAS PubMed.
- (a) S. B. Park and H. Alper, Org. Lett., 2003, 5, 3209 CrossRef CAS PubMed; (b) Z. H. Shao and H. B. Zhang, Chin. J. Org. Chem., 2005, 25, 282 CAS; (c) L. Djakovitch, M. Wagner, C. G. Hartung, M. Beller and K. Koehler, J. Mol. Catal. A, 2004, 219, 121 CrossRef CAS; (d) B. M. Choudary, S. Madhi, N. S. Chowdari, M. L. Kantam and B. Sreedhar, J. Am. Chem. Soc., 2002, 124, 14127 CrossRef CAS PubMed; (e) S. S. Stahl, Angew. Chem., Int. Ed, 2004, 43, 3400 CrossRef CAS PubMed; (f) W. Wang, C. Xiong, J. Yang and V. J. Hruby, Tetrahedron Lett., 2001, 42, 7717 CrossRef CAS; (g) B. Vaz, R. Rosana, M. Nieto, A. I. Paniello and A. R. de Lera, Tetrahedron Lett., 2001, 42, 7409 CrossRef CAS; (h) J. M. Schomaker and T. J. Delia, J. Org. Chem., 2001, 66, 7125 CrossRef CAS PubMed; (i) K. T. Wong, T. S. Huang, Y. Lin, C. C. Wu, G. H. Lee, S. M. Peng, C. H. Chou and Y. O. Su, Org. Lett., 2002, 4, 513 CrossRef CAS PubMed; (j) K. Kamikawa, T. Watanabe, A. Daimon and M. Uemura, Tetrahedron, 2000, 56, 2325 CrossRef CAS; (k) S. Paul and J. H. Clark, Green Chem., 2003, 5, 635 RSC.
- L. Yi and J. Liebscher, Chem. Rev., 2007, 107, 133 CrossRef PubMed.
- (a) R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320 CrossRef CAS; (b) H. A. Dieck and R. F. Heck, J. Am. Chem. Soc., 1974, 96, 1133 CrossRef CAS.
- (a) T. Tu, H. Mao, C. Herbert, M. Z. Xu and K. H. Dotz, Chem. Commun., 2010, 46, 7796 RSC; (b) S. Gu, H. Xu, N. Zhang and W. Chen, Chem.–Asian J, 2010, 5, 1677 CrossRef CAS PubMed; (c) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151 RSC.
- P. D. Stevens, G. Li, J. Fan, M. Yen and Y. Gao, Chem. Commun., 2005, 4435 RSC.
- Y. Wan, H. Y. Wang, Q. F. Zhao, M. Klingstedt, O. Terasaki and D. Y. Zhao, J. Am. Chem. Soc., 2009, 131, 4541 CrossRef CAS PubMed.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 CrossRef CAS PubMed.
- H. Zhao, G. Ding, L. Xu and M. Cai, Appl. Organomet. Chem., 2011, 25, 871 CrossRef CAS.
- B. Baruwati, D. Guin and S. V. Manorama, Org. Lett., 2007, 9, 5377 CrossRef CAS PubMed.
- H. Hagiwara, K. H. Ko, T. Hoshi and T. Suzuki, Chem. Commun., 2007, 2838 RSC.
- Y. H. Chen, H. H. Hung and M. H. Huang, J. Am. Chem. Soc., 2009, 131, 9114 CrossRef CAS PubMed.
- A. Honciuc, M. Laurin, S. Albu, M. Amende, M. Sobota, R. Lynch, P. Schmuki and J. Libuda, J. Phys. Chem. C, 2010, 114, 20146 CAS.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS PubMed.
- H. Lim, M. C. Cha and J. Y. Chang, Polym. Chem., 2012, 3, 868 RSC.
- M. Seki, Synthesis, 2006, 2975 CrossRef CAS.
- R. S. Varma, K. P. Naicker and P. J. Liesen, Tetrahedron Lett., 1999, 40, 2075 CrossRef CAS.
- Z. Liu, H. Wang, C. Liu, Y. Jiang, G. Yu, X. Mu and X. Wang, Chem. Commun., 2012, 48, 7350 RSC.
- R. Hudson, C.-J. Li and A. Moores, Green Chem., 2012, 14, 622 RSC.
- (a) C. Huang, K. G. Neoh, E.-T. Kang and B. Shuter, J. Mater. Chem., 2011, 21, 16094 RSC; (b) M. Mahmoudi, H. Hofmann, B. Rothen-Rutishauser and A. Petri-Fink, Chem. Rev., 2012, 112, 2323 CrossRef CAS PubMed.
- M. Shafiee, A. R. Khosropour, I. Mohammadpoor-Baltork, M. Moghadam, S. Tangestaninejad and V. Mirkhani, Catal. Sci. Technol., 2012, 2, 2440 CAS.
- (a) M. Shafiee, A. R. Khosropour, I. Mohammadpoor-Baltork, M. Moghadam, S. Tangestaninejad, V. Mirkhani and H. R. Khavasi, Mol. Divers, 2012, 16, 727 CrossRef CAS PubMed; (b) J. Noei and A. R. Khosropour, Tetrahedron Lett., 2013, 54, 9 CrossRef CAS; (c) M. Shafiee, A. R. Khosropour, I. Mohammadpoor-Baltork, M. Moghadam, S. Tangestaninejad and V. Mirkhani, Tetrahedron Lett., 2012, 53, 3086 CrossRef CAS.
- (a) A. R. Khosropour, Ultrason. Sonochem., 2008, 15, 659 CrossRef CAS PubMed; (b) J. Noei and A. R. Khosropour, Ultrason. Sonochem., 2009, 16, 711 CrossRef CAS PubMed.
- A. Landarani Isfahani, I. Mohammadpoor-Baltork, V. Mirkhani, A. R. Khosropour, M. Moghadam, S. Tangestaninejad and R. Kia, Adv. Synth. Catal., 2013, 355, 957 CrossRef.
- A. Schatz, M. Hager and O. Reiser, Adv. Funct. Mater., 2009, 19, 2109 CrossRef.
- J. F. Wei, J. Jiao, J. J. Feng, J. Lv, X. R. Zhang, X. Y. Shi and Z. G. Chen, J. Org. Chem., 2009, 74, 6283 CrossRef CAS PubMed.
- J. Wang, B. Xu, H. Sun and G. Song, Tetrahedron Lett., 2013, 54, 238 CrossRef CAS.
- A. Taher, J. B. Kim, J. Y. Jung, W. S. Ahn and M. J. Jin, Synlett, 2009, 2477 CAS.
- J. Y. Jung, J. B. Kim, A. Taher and M. J. Jin, Bull. Korean Chem. Soc., 2009, 30, 3082 CrossRef CAS.
- W. Li, B. Zhang, X. Li, H. Zhang and Q. Zhang, Appl. Catal., A, 2013, 459, 65 CrossRef CAS.
- X. Jin, K. Zhang, J. Sun, J. Wang, Zh. Dong and R. Li, Catal. Commun., 2012, 26, 199 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |