
Gold nanoparticles supported on TiO2–Ni as catalysts for hydrogen purification via water–gas shift reaction
Abstract
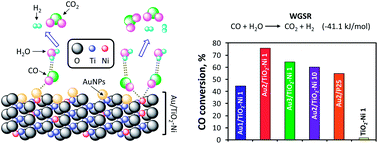
Gold nanoparticles deposited on TiO2–Ni prepared by sol–gel process catalyses the CO oxidation to hydrogen purification. Gold-catalysts were characterized by UV-Vis and Raman spectroscopies, X-ray diffraction, H2-TPR, N2 physisorption, HRTEM and STEM-HAADF microscopies. These catalysts were applied in the water–gas shift reaction at temperatures from 30 to 300 °C and this reaction was studied by DRIFTS to understand the catalytic surface phenomena. The best CO conversion was showed by doped Au/TiO2–Ni(1) with regard to Au/TiO2 sol–gel and Au/TiO2–P25 catalysts. DRIFTS confirm the strong and favorable effect of doping nickel ions into the reducible TiO2 framework. Nickel contents from 1 to 10% enhance the WGS reaction in contrast to the undoped catalyst. Ni-doped TiO2 support was practically inert for WGS reaction. These gold catalysts present significant activity in the water–gas shift reaction that allows purification of hydrogen from industrial sources at low-temperature.
 Please wait while we load your content...
Please wait while we load your content...

