
A targeted strategy for ingredients analysis and enrichment by mass-based preparative LC method: application to three isomeric C21 steroids from Marsdenia tenacissima†
Abstract
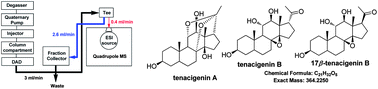
This paper proposed a strategy to acquire specified ingredients in small amounts from a complicated herbal extract by combination of simple Sephadex LH-20 column chromatography and a mass-based preparative liquid chromatography (LC) method. The developed method was applied to the analysis and enrichment of tenacigenin A, tenacigenin B, and 17β-tenacigenin B, which lack any significant chromophores from Marsdenia tenacissima. A chloroform extract of M. tenacissima was initially subjected to a Sephadex LH-20 column to yield two fractions. From 20 mg of the smaller molecular weight fraction, 3.0 mg tenacigenin A, 2.0 mg tenacigenin B and 3.0 mg 17β-tenacigenin B were successfully separated via preparative-LC employing mass-based fraction collection. In contrast to the active splitter system previously applied, the modified purification system via a tee was economic with a high convenience. The purities of the three obtained compounds were all above 90% as determined by LC coupled with evaporative light scattering detection (ELSD), and their structures were identified by 1D NMR spectra. The results demonstrated that the proposed strategy may well be an efficient and environmentally friendly technique for systematic isolation of bioactive components from traditional medicinal herbs.
 Please wait while we load your content...
Please wait while we load your content...

