DOI:
10.1039/C3RA45709E
(Paper)
RSC Adv., 2014,
4, 6738-6745
Ciprofloxacin degradation from aqueous solution by Fenton oxidation: reaction kinetics and degradation mechanisms†
Received
10th October 2013
, Accepted 2nd January 2014
First published on 6th January 2014
Abstract
Pharmaceutical wastewater from a large number of manufacturing units is extremely contaminated by ciprofloxacin (CIP), an antibiotic drug. In this work, aqueous CIP solution was treated by Fenton oxidation (FO). The effects of typical process parameters on drug mineralization have been reported. The optimal Fe2+/H2O2 molar ratio of 0.125 and pH of 3.5 were determined with 15 mg L−1 initial CIP at 25 °C temperature. Maximum CIP, COD and TOC removal of 74.4, 47.1 and 37.9% were obtained under the optimal conditions. The mean oxidation number of carbon determined in terms of COD and TOC values was in accordance with that from the oxidation number of individual carbon atom. The concentration of hydroxyl radicals was measured using the N,N-dimethyl phenyl hydrazine method using dimethyl sulphoxide as a probe. Thirteen fragments appeared in the mass spectra and the proposed mechanism explored the routes of daughter ion formation. The cleavage of the piperazine ring was more effective in CIP oxidation due to high nucleophilic character of lone pair of electrons present on the nitrogen atom. A simple 2nd order kinetic model was proposed for the oxidation of CIP and degradation products (DPs) with respect to OH˙ concentration. The rate constants of 3.13 × 103, 4.89 × 103 M−1 s−1 were estimated for CIP and DPs. The initial concentration of OH˙ was found to be 11.67 μM.
Introduction
In India, annually around 0.33 million tons of pharmaceutical waste is generated. For most of the cases, the contaminated water is disposed of into the receiving stream without suitable treatment or the water treatment facilities are not equipped to treat/filter out pharmaceuticals.1 The main reason is very high treatment cost. The concentration of pharmaceuticals in disposed water from a production unit of 90 bulk drugs in Patancheru, near Hyderabad, India, is reported to be the highest levels of pharmaceuticals in any effluent. The concentration of ciprofloxacin (CIP) was around 1000 times above the toxicity level to some bacteria. In continuation of the earlier work,1 it is reported that even a 0.2% (v/v) of this effluent could notably reduce the growth rate of tadpoles and the underlying toxicity is still unknown.2 Some pharmaceuticals can have biological activity on animals and bacteria well below the concentration that are usually used in safety tests.3 CIP is a synthetic antibiotic drug of second-generation fluoroquinolones class. It kills bacteria by interfering with the enzymes that cause DNA to rewind, which stops synthesis of DNA and protein.
Proper removal of CIP from aqueous streams has an important role in the prevention of diseases both in humans and animals. Fenton oxidation (FO) is known to be very effective advanced oxidation processes (AOPs) for the treatment of pharmaceutical wastewater.4 The primary reactions occurring in FO of organics are shown in eqn (1)–(4). R˙ indicates alkyl free radical.5
| | |
Fe2+ + H2O2 → Fe3+ + OH− + OH˙
| (1) |
| | |
Fe2+ + OH˙ → Fe3+ + OH−
| (2) |
| | |
R˙ + Fe3+ → R+ + Fe2+
| (4) |
Several in vitro experiments confirm that Fe2+ has the capacity to reduce molecular oxygen to superoxide radical for the production of the hydroxyl radical (OH˙) (eqn (5)).6
| | |
Fe2+ + O2 → Fe3+ + O2˙−
| (5) |
The overall reaction of the combined steps eqn (1) and (5) is called Haber–Weiss reaction.
| | |
O2˙− + H2O2 → O2 + OH˙ + OH−
| (6) |
In general, OH˙ react with the organic pollutants (RH) by the abstraction of H atom from C–H, N–H, or O–H bonds and by the addition of C–C bonds to the conjugate aromatic rings. The general rate equation of the key organic molecule can be written as eqn (7).
| |
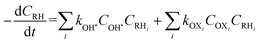 | (7) |
OXi represent other oxidants than OH˙ such as ferryl [Fe(IV)O]2+ or ˙OOH. Most of the studies assume that OH˙ formation and disappearance rate are instantaneous.7 However, it is prudent to consider [OH˙] at higher pollutant concentration.8 Investigation on [OH˙] estimation and its inclusion to the kinetics of FO for the degradation of pharmaceutical compounds still lack in literature.
Determination of OH˙ includes electron spin resonance spectroscopy in which the electron paramagnetic resonance spectrum of a spin adduct derivative is measured.9 This method is less sensitive and difficult to employ readily to acquire quantitative estimation of OH˙ as unstable OH˙-adduct is formed. OH˙ can be measured from the concentration of hydroxylated products formed with aromatic compounds such as phenol, benzoic acid and salicylic acid.10 But the problems associated in OH˙ determination in AOPs include: (i) multiple reactions, (ii) secondary generation of superoxide, (iii) limited solubility adduct and, (iv) formation of iron (+2, +3)-salicylic acid complex that hinders OH˙ formation in Fenton and Fenton-like reactions.10
To overcome these limitations, dimethyl sulphoxide (DMSO) as the chemical probe can be used for OH˙ determination. It is based on the reaction between OH˙ with DMSO to produce formaldehyde (HCHO). DMSO is highly water soluble and could trap most hydroxyl radicals generated in AOP's. It does not form complexes with iron or other metals ions in FO.11,12 Moreover, limited insight has been provided to understand the degradation mechanism of CIP. Destruction of CIP from aqueous solution is investigated using FO in this work. The influence of pH, reaction time, Fe2+ to H2O2 molar ratio on TOC, COD decay and mean oxidation number of carbon (MONC) variation have been studied. A mechanism of CIP oxidation is proposed and supported by the results obtained in LC-MS spectra. An oxidative kinetic equation involving OH˙ for [CIP] as well as for degradation products [DPs] was developed. The concentration of OH˙ was determined using DMSO as a chemical probe.
Material and methods
Reagents
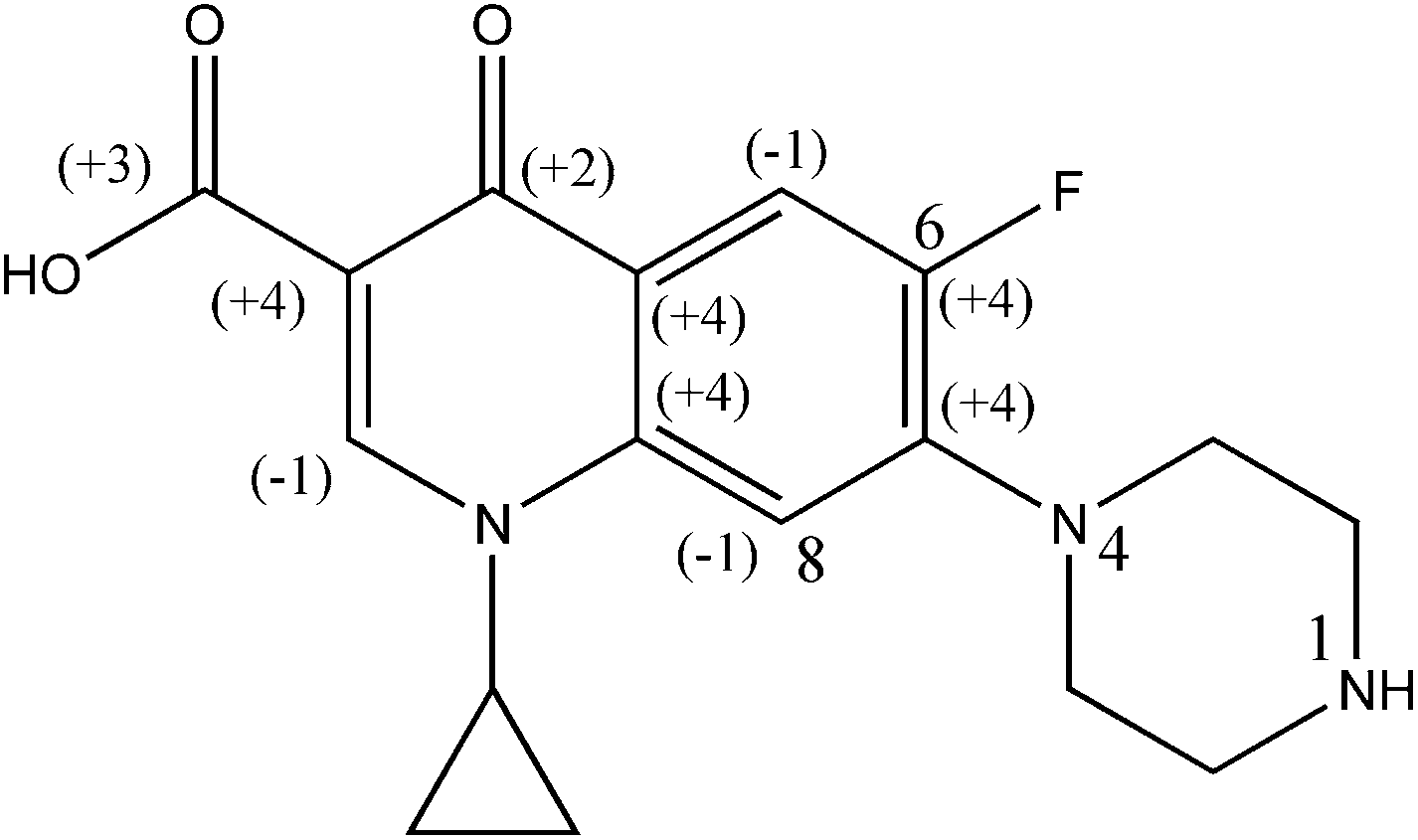
HPLC grade CIP (purity >98%, w/w) and 2,4-di-nitrophenyl hydrazine (DNPH) (purity >98%, w/w) were procured from M/s Sigma Aldrich Chemical Ltd. (USA). The chemical structure of CIP is illustrated in Fig. 1. HPLC grade of methanol (purity 98%, v/v), ferrous ammonium sulphate hexahydrate (99% purity, w/w) and sulfuric acid (98% purity, w/w), H2O2 (50% purity, v/v), Ag2SO4 (purity >98%, w/w), K2Cr2O7 (purity >98%, w/w), di-potassium hydrogen orthophosphate (98–100% purity, w/w) and DMSO (purity >96–99%, w/w), were obtained from M/s Merck Specialties Pvt. Ltd. (India). Milli-Q water (Model: Elix-3, USA) was used in preparation of reagents and drug solutions.
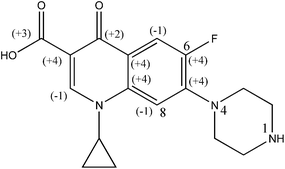 |
| | Fig. 1 Chemical structure of CIP drug [1-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-quinoline-3-carboxylic acid]. | |
Experimental
Fenton experiment. One litre capacity cylindrical (∅ 105 mm) borosilicate vessel was used as the batch reactor system. The experiment was performed at room temperature (23–25 °C) with 400 mL drug solution. The initial concentration of drug was chosen based on the maximum reported concentration in literature.13 pH of the solution was adjusted using 0.05 N H2SO4 prior to addition of Fe2+ catalyst. Solution pH was measured using a precision pH meter of M/s Eutech instruments (Model: pH/ion 510, Oaklon, Japan). Predetermined amount of Fe(NH4)2(SO4)2·6H2O was then added and mixed for 5 min at 260 rpm for better homogeneity.14 H2O2 was then added. The agitation was continued at the same speed using a magnetic stirrer (stirrer bar: length 40 mm, ∅ 0.8 mm) of M/s Tarson, Kolkata (Model: Spinot 6020). Samples were taken out at different time intervals and 0.1 N NaOH was immediately to stop the reaction at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v). Addition of NaOH increased the solution pH around 12.7. Sludge formed was separated by centrifugation at 1600 rpm for 30 min. The clear supernatant was analyzed for pH, drug concentration, COD and TOC. COD analysis was carried out heating the solution at 70 °C preceded by sludge separation.
:1 (v/v). Addition of NaOH increased the solution pH around 12.7. Sludge formed was separated by centrifugation at 1600 rpm for 30 min. The clear supernatant was analyzed for pH, drug concentration, COD and TOC. COD analysis was carried out heating the solution at 70 °C preceded by sludge separation.
Derivatization procedure. Fenton experiments were performed in duplicate under similar conditions and the second test was used for determination of OH˙ concentration. Sample was added into glass vial with previously added DMSO reagent (250 mM) at 1:0.4 (v/v). It was then mixed with 5 mL 2,4-di-nitrophenylhydrazine (DNPH)–phosphate buffer reagent. It was prepared by mixing phosphate buffer of 2.5 mL at pH 4 with 0.2 mL DNPH (6 mM in ethanol) and diluted to 5 mL with DI water. Hydrazone colored derivative formed by the reaction between HCHO and DNPH is shown in Fig. S1 of the ESI.† The reaction mixture was analyzed by LC-UV at fixed wavelength of 365 nm at room temperature. An eluent phase of 40:60 (v/v) water–methanol at 0.5 mL min−1 was employed. The retention time of the HCHO–DNPH colored derivative under these conditions was found as 7.8 min. The reactions involved for the formation of HCHO using DMSO and OH˙ are shown through eqn (8)–(11). The amount of HCHO is formed at the stoichiometric ratio of 1:2.17 with respect to OH˙.11| | |
2(CH3)2SO + 2OH˙ → 2CH3SO2H + 2CH˙3
| (8) |
| | |
2CH˙3 + 2H–R → 2CH4 + 2R˙
| (9) |
| | |
2CH˙3 + 2O2 → 2CH3OO˙
| (10) |
| | |
2CH3OO˙ → HCHO + CH3OH + O2
| (11) |
Analytical methods
High performance liquid chromatography (HPLC). A 20 μL sample was injected directly into a C18 HPLC column (150 mm length, ∅ 3.5 mm) for the determination of concentration of CIP. HPLC (Model: 26462) of M/s Shimadzu (Japan) equipped with UV-visible detector was employed for the chromatographic measurement. Acetonitrile (98% purity, v/v), water and tri-ethylamine (98% purity, v/v) at the flow rate of 1 mL min−1 (20:80:0.1 v/v/v) was used as the mobile phase. pH of the mobile phase was adjusted to 3.0 using 5% (v/v) o-phosphoric acid. The suspended particle appeared was removed by filtration using 0.45 μm cellulose acetate filter. The scanning was performed at fixed wavelength of 280 nm.
Liquid chromatography-mass spectroscopy (LC-MS). HPLC method outlined above was adopted for subsequent sample introduction to MS detector. The chromatographic separation was performed on a YMC Hydrosphere C18 150 mm × 4.6 mm (5 μm particle size) reverse phase analytical column (Wilmington, NC, US) following a YMC Hydrosphere 10 mm × 4 mm (5 μm particle size) guard column. The mobile phase flow rate was 0.8 mL min−1. H2O and acetonitrile with 0.1% (v/v) formic acid was employed as the mobile phase. A linear gradient of 95 to 50% water for 15 min was used. A 10 μL of both sample and calibration solution were injected into the column using an AS 3000 auto injector (Thermo Finnigan, USA). Atmospheric pressure chemical ionization method in positive ion mode over the mass range of 100 to 500 amu was adopted. N2 at a flow rate of 400 L h−1 for drying and 150 L h−1 for sheathing were purged. The source temperature at 120 °C and a cone voltage of 25 V were maintained to determine the m/z ratio of the parent ion and isotope. A cone voltage of 75 V was used for the fragmentation of the daughter ions. The electrospray source voltage was held at 3.5 kV and the temperature was maintained at 200 °C.
TOC and COD analysis. Total organic carbon (TOC) analyzer of M/s O.I. Analytical (Model: 1030C Aurora, USA) was employed for determination of TOC before and after experiments. Non-dispersive infrared method was adopted for the detection. Chemical oxygen demand (COD) was determined by scanning the sample at 360 nm wavelength using HACH DRB 200 digester following the protocol recommended by the manufacturer.
Results and discussion
Optimal Fe2+/H2O2 molar ratio
The dose of Fe2+ and H2O2 determine the operational cost as well as the efficiency of degradation of contaminant in FO. The concentration of Fe2+ drops quickly with the progress of FO (eqn (1)). It is consequently balanced by the formation of Fe2+ through reduction of Fe3+ (eqn (12)) and Fe2+ concentration reaches at the steady state.| | |
Fe3+ + H2O2 → Fe2+ + OOH˙ + H+
| (12) |
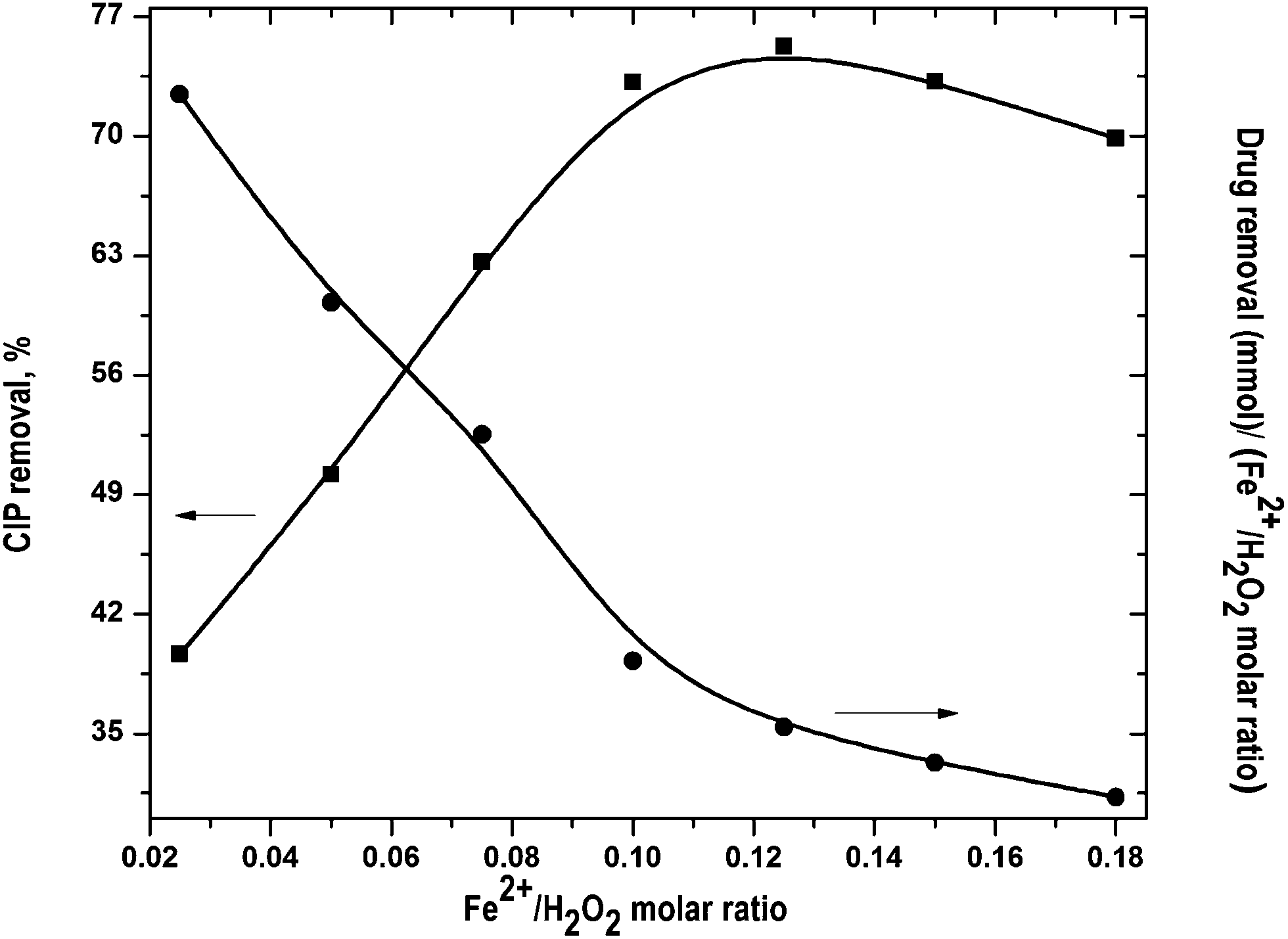
Optimal Fe2+/H2O2 molar ratio was found out by varying it in the range from 0.05 to 0.18 and H2O2 dose of 10 mM was used in all experiments. The change in [OH˙] can be divided into two phases. At the initial phase, OH˙ radicals production is faster. It resulted in higher CIP degradation (Fig. S2 of the ESI†). In the second phase, [OH˙] decreased even though CIP oxidation mostly occurred within the 1st stage.15 It was primarily due to progressive depletion of H2O2.
The results at the end of the run are shown in Fig. 2. CIP removal was increased with increase in Fe2+/H2O2 molar ratio and maximum drug removal of 74.4% was obtained at 0.125. CIP removal decreased with further rise of Fe2+/H2O2 ratio. Generally, the rate of OH˙ formation increases with rise of Fe2+/H2O2. However, excess H2O2 with respect to Fe2+ can directly oxidize Fe2+ to Fe3+. It reduces the production rate of OH˙ and drug removal fell down.16,17 CIP removal per unit Fe2+/H2O2 molar ratio dropped almost linearly (Fig. 2). It suggests that FO was more proficient in consumption of Fe2+ and H2O2 for drug degradation at lower dose. At Fe2+/H2O2 >0.125, CIP removal dropped a bit. It implies that the optimal Fe2+/H2O2 ratio was around 0.125 in terms of percentage CIP removal.
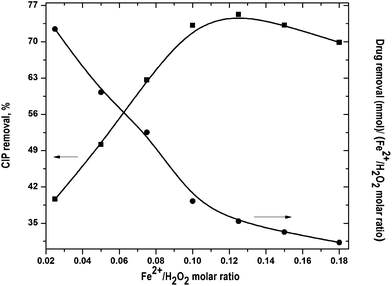 |
| | Fig. 2 Effect of Fe2+/H2O2 molar ratio on CIP degradation. Experimental conditions: [CIP]0 = 15 mg L−1, reaction time = 45 min, pH = 3.5 and temperature = 25 °C. | |
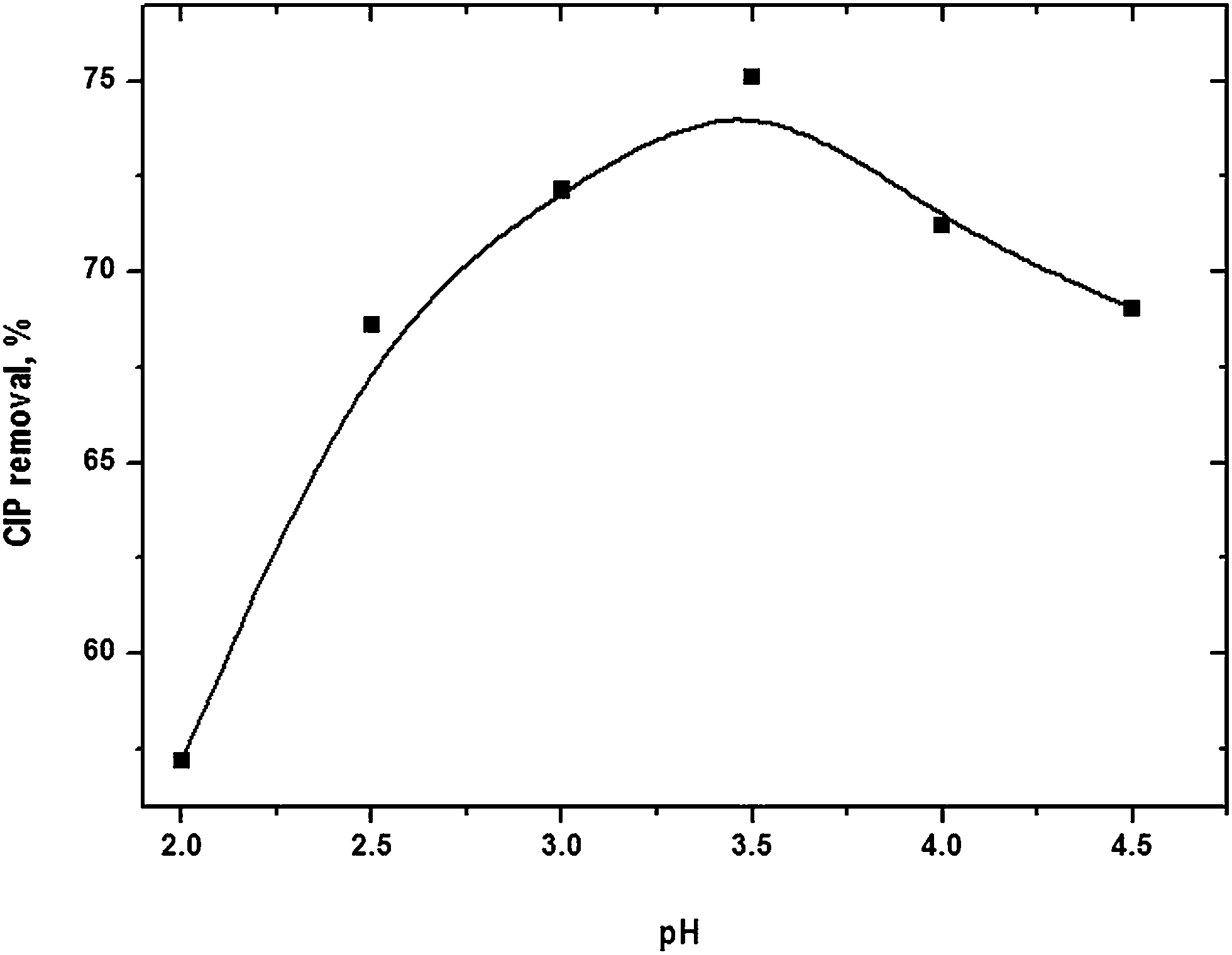
Optimal pH
pH of the solution has notable influence on FO. It controls the production rate of hydroxyl radical and the nature of iron species. pH was varied in the range from 2 to 4.5 (Fig. S3 of the ESI†). The dynamics of drug removal followed similar trend at different pH. Removal efficiency gradually increased with rise of pH and it reached a maximum value of 74.4% at pH 3.5 (Fig. 3). Further pH elevation decreased drug removal. OH˙ could be consumed by the scavenging effects of H+ (eqn (13)) at pH < 3.5 which would limit the degradation rate.18 It might also be possible that H2O2 was stable by acquiring a proton to form an oxonium ion (eqn (14)) at lower pH.19
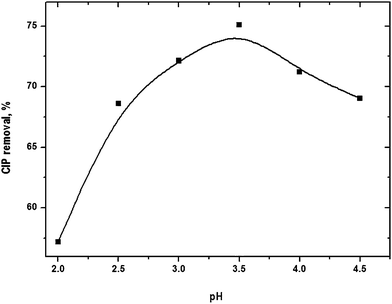 |
| | Fig. 3 CIP degradation as a function of solution pH. Experimental conditions: [CIP]0 = 15 mg L−1, Fe2+/H2O2 molar ratio = 0.125, reaction time = 45 min and temperature = 25 °C. | |
The concentration iron used in Fenton experiment was lower than the theoretical solubility of Fe(II) and Fe(III).20,21 [Fe2+] is more than 99.9% at pH < 4.22 However, brownish appearance of the solution was visually noted with addition of CIP into Fenton reagent and it transformed into steady particle form in about 10 min. It implies that probably iron-organo complexes of lower solubility were formed. It reduced the total available iron, primarily Fe2+, and CIP in solution for FO. Sludge may be converted to more stable solid modifications such as α-FeOOH(s) and α-Fe2O3(s) with progress of the reaction. Indeed, highly heterogeneous nature of the iron sludge is formed in FO.23,24 The results obtained at different pH indicate that the optimal pH of CIP removal was around 3.5.
Comparative CIP, COD and TOC removal at optimal condition
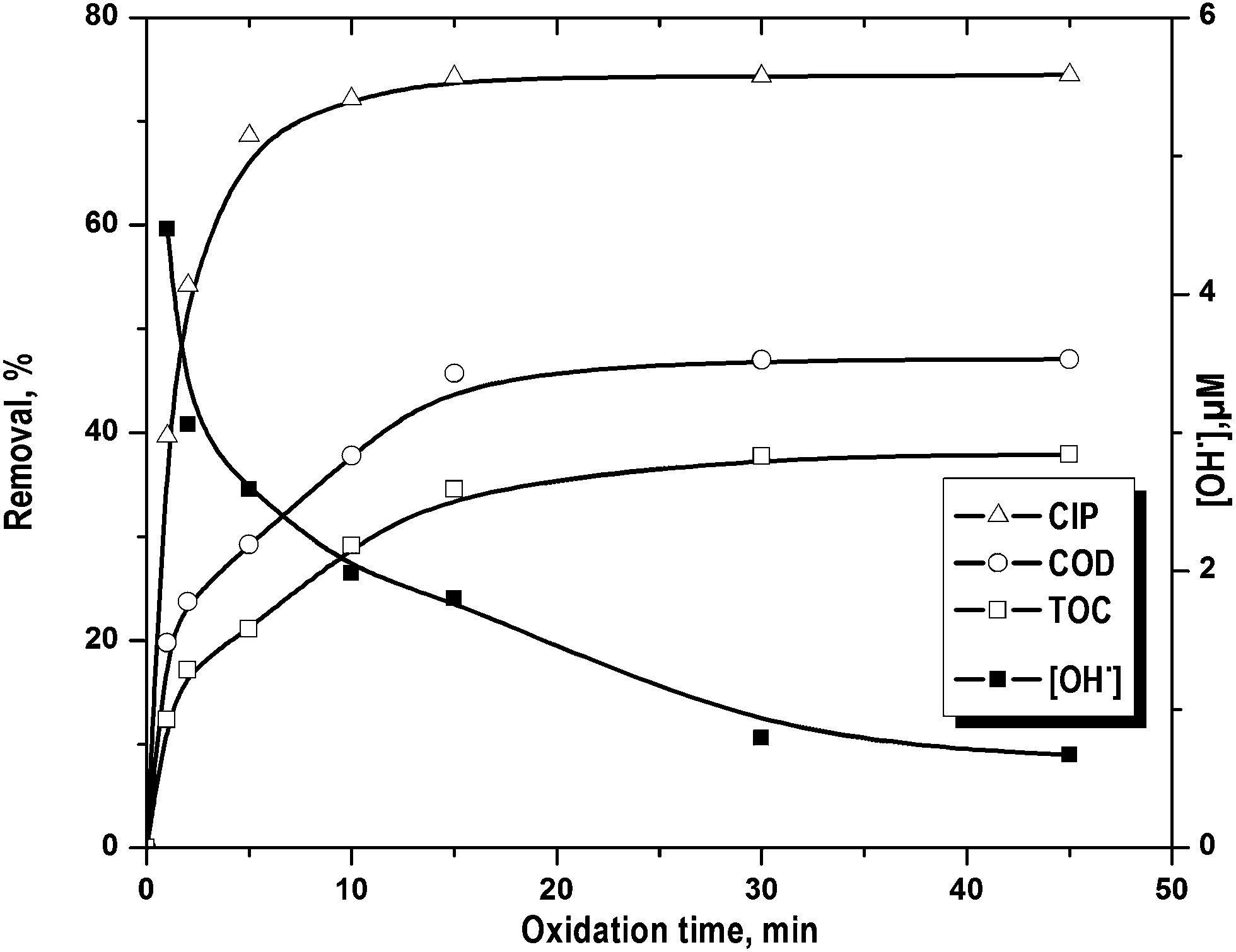
CIP removal showed two distinct rate periods (Fig. 4) i.e. initial faster drug removal followed by an almost constant rate even though there was considerable amount of unreacted CIP. Percentage COD and TOC removal increased gradually with the reaction time. On the other hand, the change in CIP decomposition was <5.8% after 5 min of FO (Fig. 4). This is in accordance to the change in OH˙ concentration (Fig. 4). It dropped quickly within first 5 min of FO and then it decreased gradually. The rate of formation of OH˙ decreased as a net result of Fe2+ catalytic effect reduction, OH˙ scavenging by H+ (eqn (13)) and H2O2 auto decomposition (eqn (14)).25 The high initial rate of OH˙ formation was resulted in higher initial rate of CIP (till 5 min), COD and TOC (till 15 min) removal. Maximum COD and TOC removal were found as 47.1 and 37.9% in 15 min against the drug removal of 74.4%. After that, there was no notable effect of treatment time on CIP, COD and TOC removal.
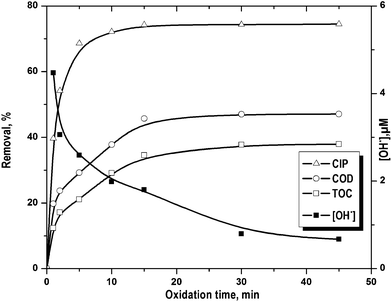 |
| | Fig. 4 Removal of CIP, COD and TOC along with the variation hydroxyl radical concentration with the progress reaction time. Experimental conditions: [CIP]0 = 15 mg L−1, Fe2+/H2O2 molar ratio = 0.125, pH = 3.5 and temperature = 25 °C. | |
Sludge formed during FO was separated by filtration using 0.45 μm cellulose ester filter. It was then washed in distilled water and dissolved in conc. H2SO4 solution. Amount of CIP adsorbed on sludge was determined after 45 min of FO under the optimal condition (Fe2+/H2O2 molar ratio 0.125 and pH 3.5). About 3, 4 and 6% CIP, COD and TOC reduction was observed due to sludge appearance. It implies that CIP adsorption on iron sludge would not affect the kinetics of CIP oxidation. Stable CIP (or intermediates)–iron complex was formed which was resistant to FO. It is in line with the steady appearance of iron–organic sludge.
Mean oxidation number of carbon
The mean oxidation number of carbon (MONC) reveals the experimental errors incurred in COD and TOC measurements. Chemically, the MONC must lie in the range from −4 to 4 with respect to the simplest organic compound, CH4. The MONC for a single organic molecule containing ‘n’ number of carbon atoms can be represented as in eqn (15). ONi indicates the oxidation number of ith carbon atom (eqn (15)).26 The oxidation number of the individual carbon atom is provided in Fig. 1 in round parenthesis for the CIP molecule. An oxidation number of 0, +1, +0.5, +2 were considered for the C–C σ- and π-bonds; C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C σ- and π-bonds and; CO σ- and π-bonds, respectively.27 It gave net ‘zero’ oxidation number of carbons in both piperazine ring and cyclopropyl group.
C σ- and π-bonds and; CO σ- and π-bonds, respectively.27 It gave net ‘zero’ oxidation number of carbons in both piperazine ring and cyclopropyl group.| |
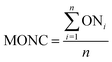 | (15) |
COD and TOC values also can be combined together to compute the MONC irrespective to the number of organic molecule present in the sample (eqn (16)).28
| |
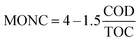 | (16) |
COD and TOC are expressed in mg L
−1. Iron sludge formed during FO was removed preceded by Fe
2+ oxidation at high pH (
ref. 29) and residual OH˙ and H
2O
2 were destroyed by heating the samples to avoid the interference with inorganic COD. The experimentally determined COD value was therefore readily employed for the estimation of MONC. The results are shown in
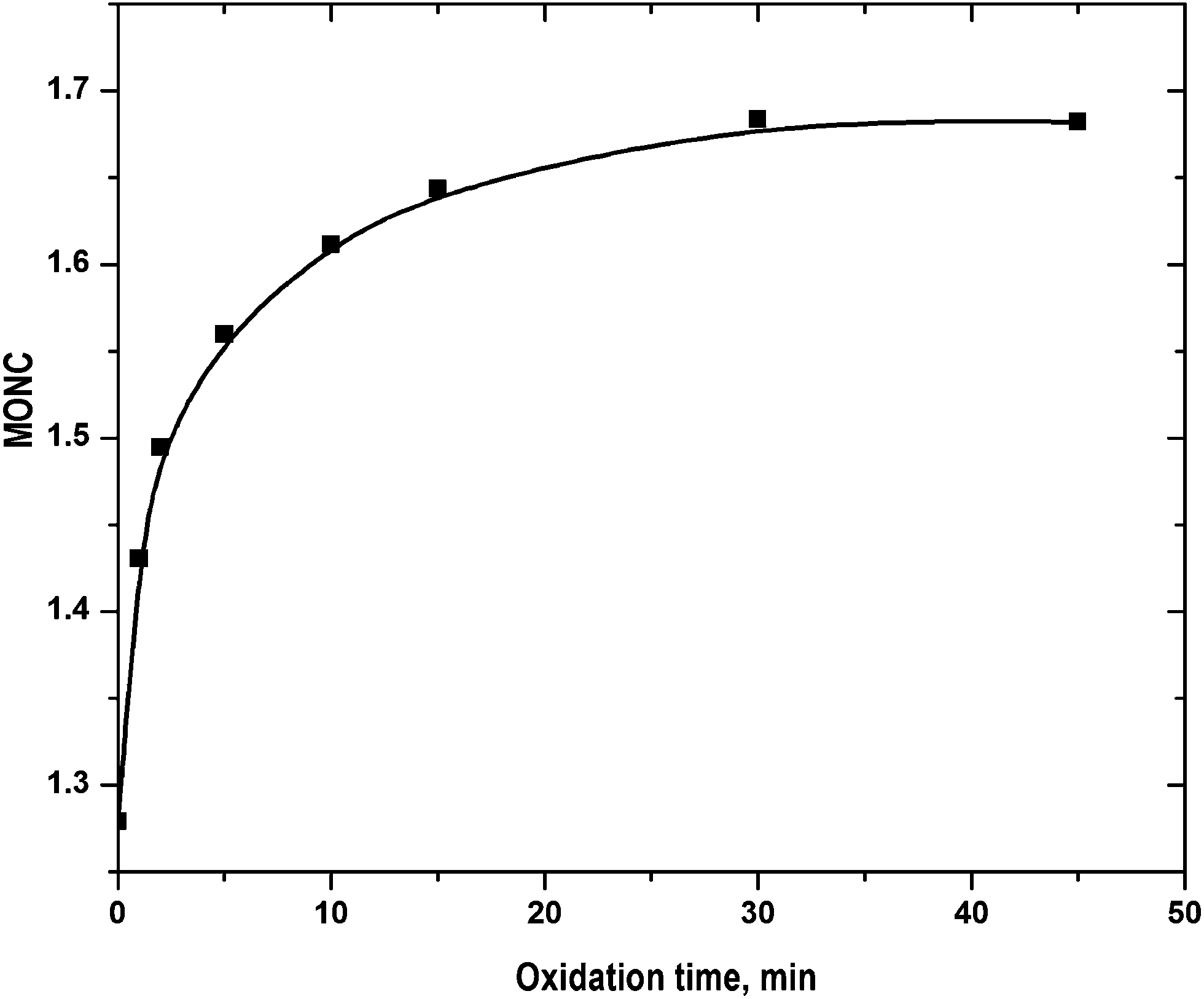
Fig. 5. The initial MONC values estimated using
eqn (15) and
(16), were of 1.17 and 1.28. It implies that COD and TOC were determined with reasonable accuracy. The MONC increase was faster at <5 min and then it increased gradually. The MONC raised to 1.68 in 45 min of FO. The result can be corroborated by the refractory nature of the quinolone moiety. It contains 9 carbon atoms out of 17 in CIP molecule.
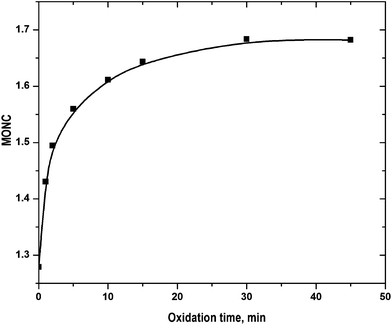 |
| | Fig. 5 Variation of MONC with progress of reaction: experimental conditions: [CIP]0 = 15 mg L−1, Fe2+/H2O2 molar ratio = 0.125, pH = 3.5 and temperature = 25 °C. | |
Proposed mechanisms of CIP degradation
N4 in piperazine ring of fluoroquinolone compounds is typically the specific site of OH˙ attack. N1 is likely less reactive than N4 because of its weaker basicity.30 Two strong electron-withdrawing nitrogen substituents i.e. fluorine and –COOH groups are therein the aromatic ring (Fig. 1). On the other way it indicates that fluoroquinolone ring is less electron-donating because of highly electron-withdrawing fluorine substituent.22 In general, the loss of carboxyl moiety ([M + H-44]+) and fluoride ([M + H-22]+), are the typical fragmentation pathways of fluoroquinolone.31 The appearance of such fragmentations in the MSn spectra gives some hints for the subtraction of different moieties based on their mass losses such as 44 for carboxyl group.
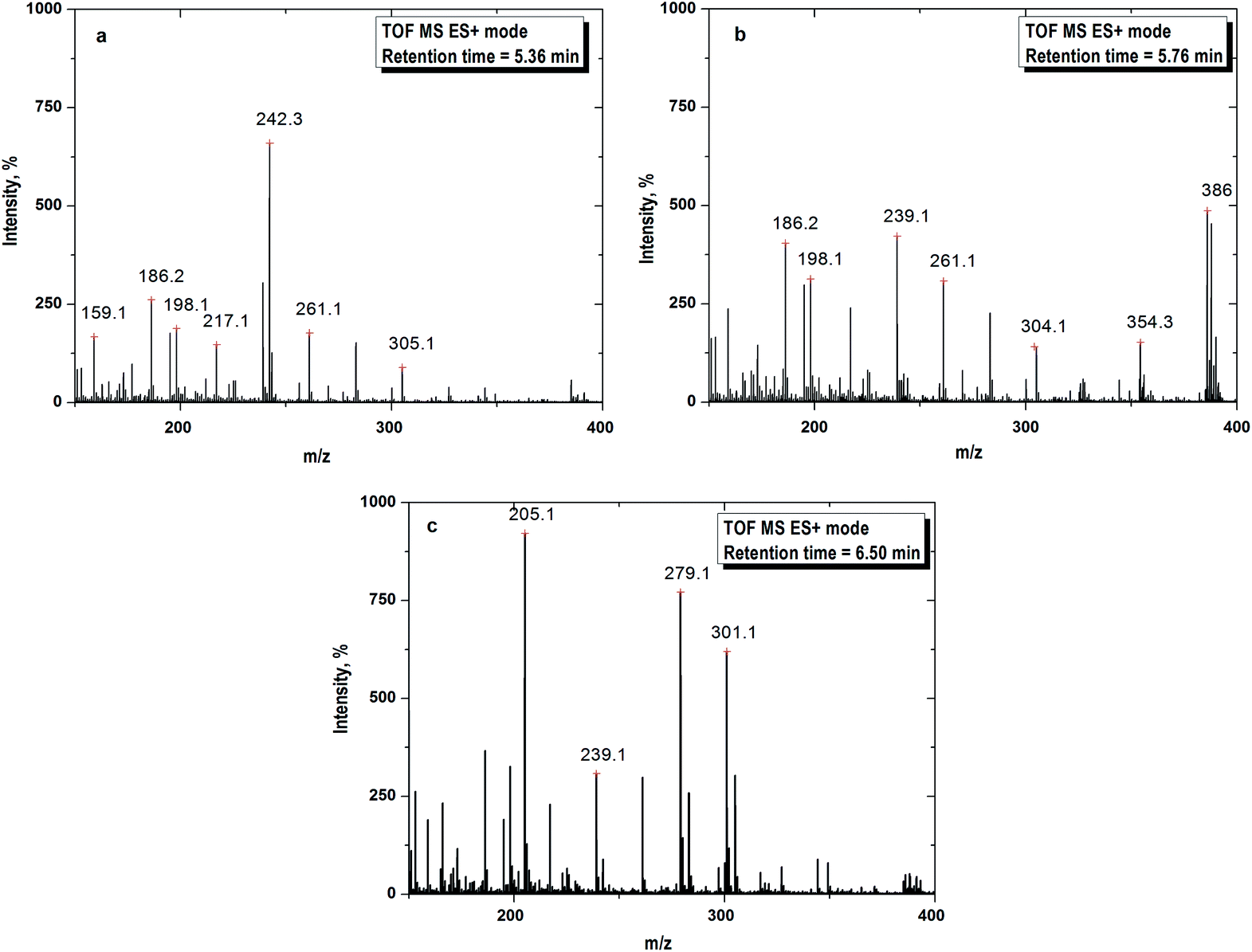
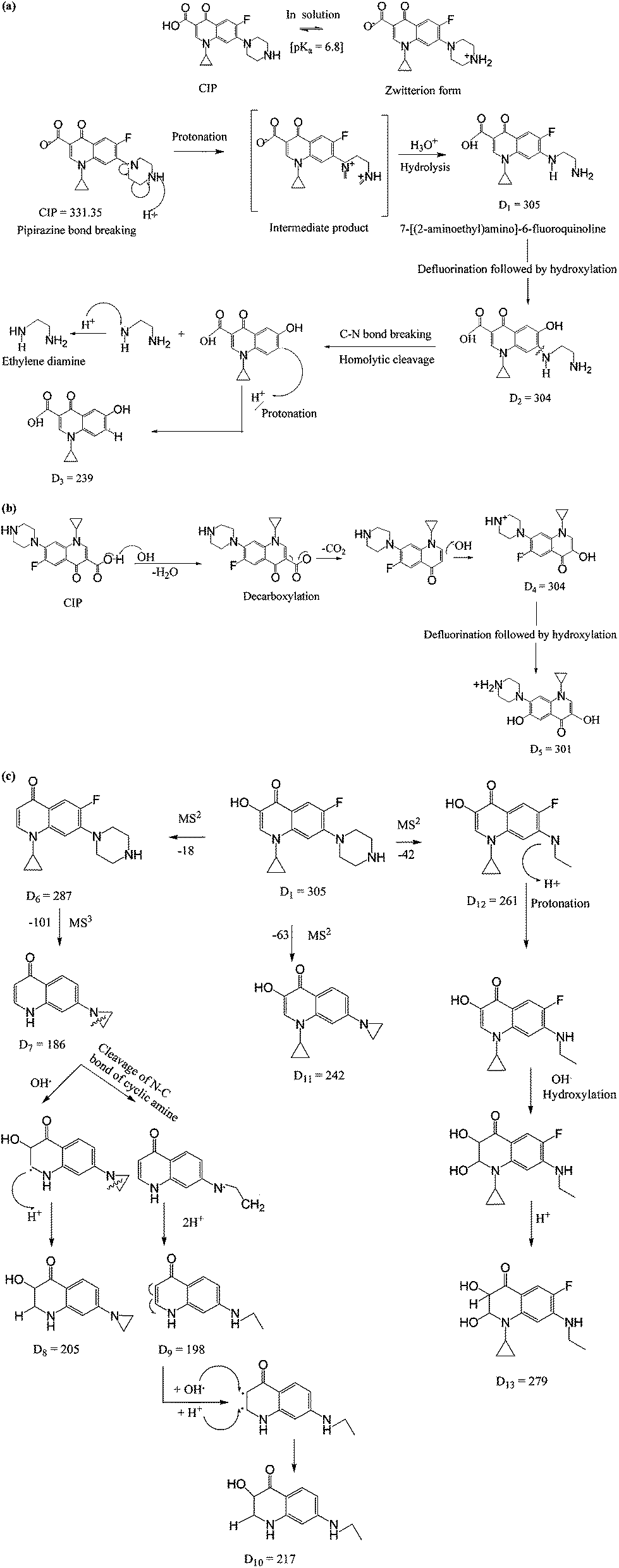
The mass spectra are depicted in Fig. 6. The formation of daughter ions could follow two pathways for the degradation of CIP i.e. piperazine moiety degradation and decarboxylation. The possible routes of oxidation of CIP molecule are illustrated in Fig. 7. The peaks in the mass spectra with m/z of 305, 186, 198, 217, 242 and 261 were for the retention time of 5.36 min (Fig. 6a). All the degradation products are denoted with the symbol ‘D’ following the integer to count the number of fragments. D1 with m/z 305 has the same specific mass of 7-[(2-amino-ethyl)amino]-6-fluoroquinoline formed by the loss of the piperazine ring (Fig. 7a–c) under acidic conditions. It appeared with the cleavage of piperazine ring via an unstable intermediate product as shown in Fig. 7a. The nitrogen atom of piperazine ring with a lone pair electron, abstracted the proton from the solvent at acidic pH and the C–C bond was broken.
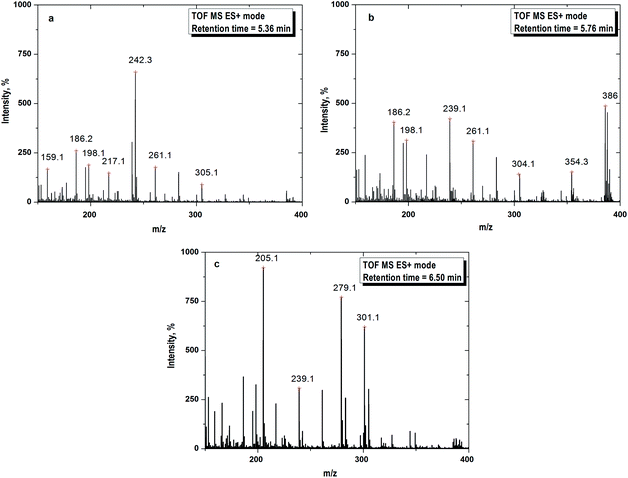 |
| | Fig. 6 (a) Mass spectra recorded at 10 min of FO for daughter ion 1 (D1) with m/z = 305. (b) Mass spectra recorded at 10 min of FO for daughter ion 2 (D2) with m/z = 304. (c) Mass spectra recorded at 10 min of FO for daughter ion 3 (D3) with m/z = 301. Experimental conditions: [CIP]0 = 15 mg L−1, Fe2+/H2O2 molar ratio = 0.125, pH = 3.5 and temperature = 25 °C. | |
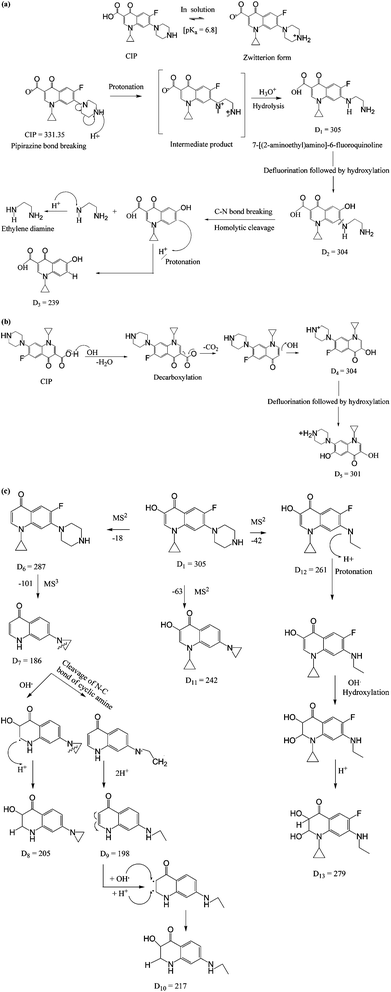 |
| | Fig. 7 Proposed mechanisms for the formation of daughter ions. (a) Mechanism 1: piperazinyl moiety degradation. (b) Mechanism 2: decarboxylation. (c) Further fragmentation of D1 with m/z 305. | |
The fragmentations with m/z of 186, 198, 239, 261, 304, 354 and 386 at 5.76 min of retention are presented in Fig. 6b. The daughter ions with m/z ratio of 304 (D2) were formed due to piperazine ring breaking along with defluorination followed by hydroxylation of D1 (Fig. 7a). D3 (m/z of 239) appeared with the loss of fluorine atom at position 6 and expulsion of ethylenediamine molecule (Fig. 7a).
Decarboxylation of CIP led to formation of D4 having the same m/z of D2. MS spectra of the compound with m/z = 301 (D5) theoretically represents a fragment of CIP which might be formed by the loss of fluorine atom (Fig. 7b). The steric effect between carbonyl and carboxylic (–COOH) groups and the presence of fluoride group could trigger such decarboxylation.
The peaks with m/z of 205, 239, 279 and 301 in the mass spectra corresponding to retention time of 6.50 min are shown in Fig. 6c. –OH group was expelled out from the quinolone moiety in presence of electron withdrawing N atom and carbonyl group. Therefore, D1 was further broken into D6 (m/z of 287) by dehydroxylation. Piperazine ring suffers an angle stress in presence of high electronegative fluorine atom. It led to formation of the three-membered cyclic amine ring (D7) (Fig. 7c). Amine group makes the quinolone moiety as an electron rich compound and it is easily targeted by OH˙ because of electrophile nature of the free radical. As a result, D8 was formed by hydroxylation. D9 (m/z 198) and D10 (m/z 217) were yielded due to cleavage of N–C bond of cyclic amine ring (Fig. 7c). D1 broken into D11 (m/z 242) on defluorination and partial piperazine moiety oxidation because of steric hindrance (Fig. 7c). A similar explanation for D12 is also applicable. The possible route for the formation of D13 with m/z 279 is shown in Fig. 7c. The pathways for the formation of daughter ions are summarized in Table S1 (ESI†). Lower TOC removal is in accordance with the proposed mechanism of CIP degradation as carbon atoms mainly remained to the quinolone moiety. The variation of MONC also gives the hint for the successive oxidation of the CIP molecule. The MONC for the individual daughter ions were varied in the range of 0.8 (D11) to 1.72 (D8) at 10 min of FO with 28% unreacted CIP against the same of 1.18 for CIP.
Kinetic model for the oxidation of CIP and degradation products
CIP molecules are targeted by OH˙ immediately upon its generation (eqn (1)) and a number of products are formed. The solution in the batch reactor was completely mixed and the reaction temperature was controlled in a narrow range (23 to 25 °C). The pH variation was insignificant during degradation experiment. It was assumed that active OH˙ was the main oxidant to cleavage the organic substances in the solution.
OH˙ radical was utilized for the oxidation of CIP (eqn (17)) and degradation products (DPs). The initial CIP concentration was quite high and OH˙ showed exponential decay with oxidation time (Fig. 4) at the optimal Fe2+/H2O2 and pH. It implies that the rate of CIP oxidation was dependent on [OH˙]. So the rate equation is expressed in terms of concentration of CIP (eqn (18)) and DPs (eqn (19)). k1 and k2 (M−1 s−1) are the second order rate constants. CIP, DPs and OH˙ in square parenthesis indicate their concentration.
| | |
CIP + OH˙ → CIPoxidised
| (17) |
| |
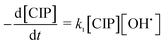 | (18) |
| |
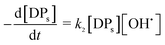 | (19) |
eqn (18) and
(19) can be combined as in
eqn (20):
| |
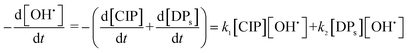 | (20) |
The TOC values of unreacted CIP and DPs were experimentally found out at different time intervals. The contribution of TOC due to unreacted CIP was determined at the same concentration of CIP. TOC of DPs was calculated by subtracting the share for unreacted CIP. An atomic number average molecule (molecular weight 218 g mol−1) was proposed from the structure of intermediates (13 nos) as in the mechanisms (Fig. 7a–c). Hence, the concentration of DPs was calculated from their TOC values. Therefore, a better estimation of initial OH˙ radical concentration 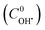 and the second order rate constant of CIP oxidation can be done. However, the uncertainly may arise from the calculation of average molecular weight of DPs.
and the second order rate constant of CIP oxidation can be done. However, the uncertainly may arise from the calculation of average molecular weight of DPs.
The least square technique was used to estimate the kinetic constants and 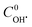 . The best fit graphics are shown in Fig. S4–S6 (ESI†). The results imply that both CIP and DPs fairly followed the 2nd order kinetics.
. The best fit graphics are shown in Fig. S4–S6 (ESI†). The results imply that both CIP and DPs fairly followed the 2nd order kinetics. 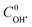 , k1 and k2 were found as 0.051 μM, 3.13 × 103 and 4.89 × 103 M−1 s−1, respectively. Higher k2 value implies that the overall reactivity of DPs towards OH˙ was more than CIP.
, k1 and k2 were found as 0.051 μM, 3.13 × 103 and 4.89 × 103 M−1 s−1, respectively. Higher k2 value implies that the overall reactivity of DPs towards OH˙ was more than CIP. 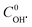 exhibited good accordance to the earlier reports. Hayashi et al.32 showed that [OH˙] in FO typically varies in the range from 1 to 0.01 μM. The average OH˙ generation and CIP degradation rates were found as 3.83 × 10−8 and 2.97 × 10−8 M s−1, respectively. Maezono et al.33 reported a close value of the average OH˙ formation rate as 9.3 × 10−10 M s−1 for the oxidation of Orange II with 60 mg L−1 at pH 3 and Fe2+/H2O2 molar ratio of 0.01 in photo-assisted Fenton process.
exhibited good accordance to the earlier reports. Hayashi et al.32 showed that [OH˙] in FO typically varies in the range from 1 to 0.01 μM. The average OH˙ generation and CIP degradation rates were found as 3.83 × 10−8 and 2.97 × 10−8 M s−1, respectively. Maezono et al.33 reported a close value of the average OH˙ formation rate as 9.3 × 10−10 M s−1 for the oxidation of Orange II with 60 mg L−1 at pH 3 and Fe2+/H2O2 molar ratio of 0.01 in photo-assisted Fenton process.
Conclusions
The following conclusions are drawn from this investigation:
• The maximum CIP removal of 74.4% was found with [CIP]0 = 15 mg L−1 at optimal pH = 3.5 and Fe2+/H2O2 = 0.125. COD and TOC removal were of 47.1 and 37.9%. A large portion of intermediates were resistant to FO.
• The residual hydroxyl radical concentration of 0.792 μM was determined using DMSO probe after 30 min of FO even with 25.7% unreacted CIP.
• The mean oxidation number of carbon increased from 1.28 to 1.68 at the optimal conditions.
• Four primary daughter ions were originated upon degradation of CIP during FO. D1 (m/z 305) and D2 (m/z 304) were produced by piperazine moiety degradation. Whereas, D3 (m/z 304) along with D4 (m/z 301) were formed by decarboxylation. The daughter ion with m/z ratio of 305 was further broken into smaller fragments.
• A 2nd order kinetic model for the cleavage of both CIP and degradation products (DPs) exhibited excellent agreement to the experimental data. The rate constant of CIP and DPs oxidation were obtained as 3.13 × 103 and 4.89 × 103 M−1 s−1. The model gave an initial hydroxyl radical concentration of 11.67 μM.
References
- D. G. J. Larsson, C. de Pedro and N. Paxeus, J. Hazard. Mater., 2007, 148, 751–755 CrossRef CAS PubMed.
- L. Gunnarsson, E. Kristiansson, C. Rutgersson, J. Sturve, J. Fick, L. Forlin and D. G. J. Larsso, Environ. Toxicol. Chem., 2009, 28(12), 2639–2647 CAS.
- H. J. H. Fenton, J. Chem. Soc., 1984, 65, 899–910 RSC.
- Y. Hu, Y. Zhang and Y. Tang, RSC Adv., 2012, 2, 6036–6041 RSC.
- S. Hyun-Seok, K. Jeehyeong and K. Zoh, Environ. Prog. Sustainable Energy, 2010, 29(4), 415–420 CrossRef.
- M. C. Dodd, A. D. Shah, U. G. Von and C. H. Huang, Environ. Prog. Sustainable Energy, 2005, 34, 7065–7076 Search PubMed.
- D. Mantzavinos, A. G. Livingstong and R. Hellenbrand, Chem. Eng. Sci., 1996, 51(18), 4219–4235 CrossRef CAS.
- R. C. Martins, A. F. Rossi and R. M. Quinta-Ferreira, J. Hazard. Mater., 2010, 180, 716–721 CrossRef CAS PubMed.
- T. E. Doll and F. H. Frimmel, Water Res., 2004, 38, 955–964 CrossRef CAS PubMed.
- G. A. Surucu Gulkaya and F. B. Dilek, J. Hazard. Mater., 2006, 136(3), 763–769 CrossRef PubMed.
- E. M. Lindsey and A. M. Tarr, Chemosphere, 2000, 41, 409–417 CrossRef.
- A. T. Ternes, J. Stuber, N. Herrmann, D. McDowell, A. Ried, M. Kampmann and B. Teiser, Water Res., 2003, 37, 1976–1982 CrossRef.
- W. Yang, N. Cicek and J. Ilg, J. Membr. Sci., 2006, 270(1–2), 201–211 CrossRef CAS PubMed.
- A. Serra, X. Dom, E. Brillas and J. Peral, J. Environ. Monit., 2011, 13, 167–174 RSC.
- N. Saadatjou, M. Taghdiri and R. Farrokhi, Iran. J. Environ. Health Sci. Eng., 2010, 7(4), 345–352 CAS.
- E. Kugelmann, R. C. Albert, G. Bringmann and U. Holzgrabe, J. Pharm. Biomed. Anal., 2011, 54, 1047–1058 CrossRef CAS PubMed.
- K. S. Shrivastaval, N. S. Rathore and A. K. Solanki, J. Pharm. Sci. Technol., 2010, 2(3), 163–170 Search PubMed.
- W. Stumm and J. J. Morgan, Aquatic Chemistry, John Wiley, New York, 2nd edn, 1981, vol. 3, ch. 2, pp. 191–195 Search PubMed.
- D. W. Kolpin, E. T. Furlong, M. T. Meyer, E. M. Thurman and H. T. Buxton, Environ. Sci. Technol., 2002, 36, 1202–1211 CrossRef CAS.
- P. Ghosh, P. Kelapure, A. N. Samanta and S. Ray, Int. J. ChemTech Res., 2012, 4(1), 116–123 CAS.
- H. Tekin, O. Bilkay, S. Selale and H. Tolga, J. Hazard. Mater., 2006, 136B, 258–265 CrossRef PubMed.
- M. Klavarioti, D. Mantzavinos and D. Kassinos, Environ. Int., 2009, 35, 402–417 CrossRef CAS PubMed.
- Z. Qiang, J. H. Chang and C. P. Huang, Water Res., 2003, 37, 1308–1319 CrossRef CAS.
- Y. Segura, F. Martinez and J. A. Melero, Appl. Catal., B, 2013, 136–137, 64–69 CrossRef CAS PubMed.
- F. Vogel, J. Harf, A. Hug and P. R. Von Rohr, Water Res., 2000, 34, 2689–2702 CrossRef CAS.
- M. Hesse, H. Meier and B. Zeeh, in Spectroscopic Methods in Organic Chemistry, Thieme, New York, 2nd edn, 2008, vol. 4, ch. 7, pp. 17–21 Search PubMed.
- Y. Zhao, M. Huang, M. X. Tang and L. Liu, J. Environ. Monit., 2010, 12, 271–279 RSC.
- H. Park, H. T. Kim and K. Barak, Eur. J. Med. Chem., 2002, 37, 443–448 CrossRef CAS.
- P. Wang, Y. L. He and C. H. Huang, Water Res., 2010, 44, 5989–5998 CrossRef CAS PubMed.
- H. Zhang and C. H. Huang, Environ. Sci. Technol., 2005, 39, 4474–4483 CrossRef CAS.
- D. Klauson, J. Babkina, K. Stepanova, M. Krichevskaya and S. Preis, Catal. Today, 2010, 151, 39–45 CrossRef CAS PubMed.
- H. Hayashi, Y. Nakajima and K. Ohta, Rep. Ind. Technol. Res. Inst., 2007, 21, 79–83 CAS.
- T. Maezono, M. Tokumura, M. Sekine and Y. Kawase, Chemosphere, 2011, 82, 1422–1430 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra45709e |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 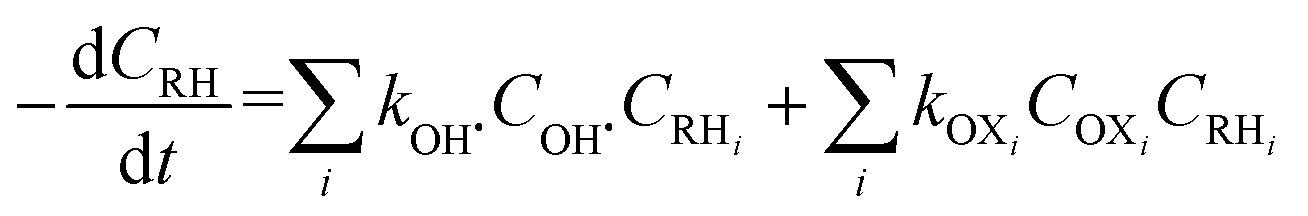
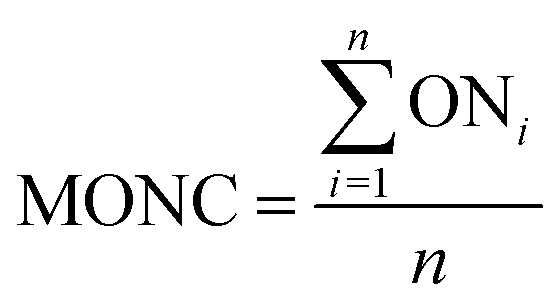

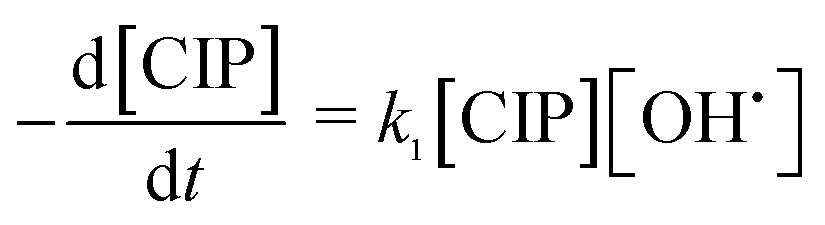
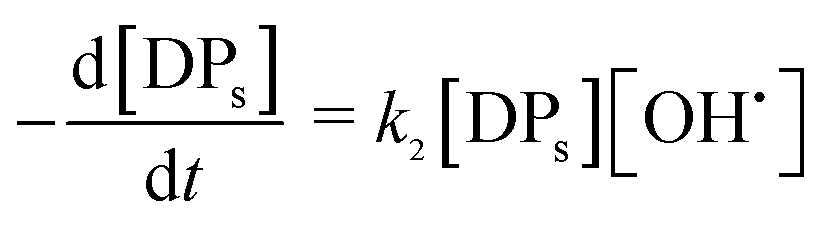
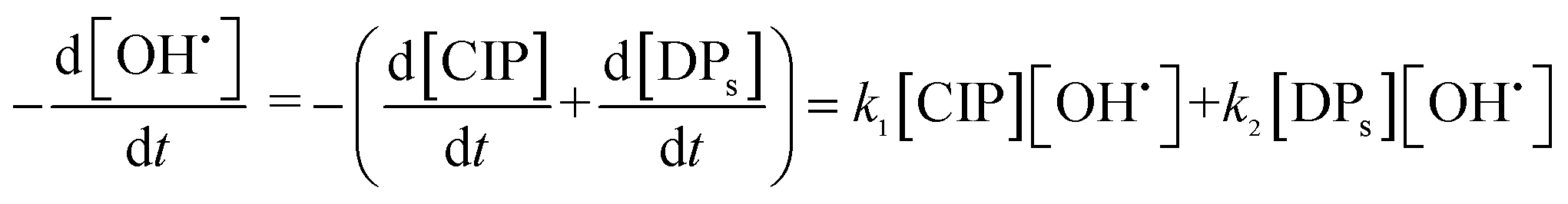
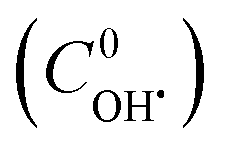 and the second order rate constant of CIP oxidation can be done. However, the uncertainly may arise from the calculation of average molecular weight of DPs.
and the second order rate constant of CIP oxidation can be done. However, the uncertainly may arise from the calculation of average molecular weight of DPs.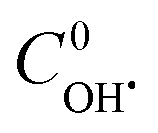 . The best fit graphics are shown in Fig. S4–S6 (ESI†). The results imply that both CIP and DPs fairly followed the 2nd order kinetics.
. The best fit graphics are shown in Fig. S4–S6 (ESI†). The results imply that both CIP and DPs fairly followed the 2nd order kinetics. 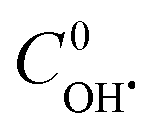 , k1 and k2 were found as 0.051 μM, 3.13 × 103 and 4.89 × 103 M−1 s−1, respectively. Higher k2 value implies that the overall reactivity of DPs towards OH˙ was more than CIP.
, k1 and k2 were found as 0.051 μM, 3.13 × 103 and 4.89 × 103 M−1 s−1, respectively. Higher k2 value implies that the overall reactivity of DPs towards OH˙ was more than CIP.  exhibited good accordance to the earlier reports. Hayashi et al.32 showed that [OH˙] in FO typically varies in the range from 1 to 0.01 μM. The average OH˙ generation and CIP degradation rates were found as 3.83 × 10−8 and 2.97 × 10−8 M s−1, respectively. Maezono et al.33 reported a close value of the average OH˙ formation rate as 9.3 × 10−10 M s−1 for the oxidation of Orange II with 60 mg L−1 at pH 3 and Fe2+/H2O2 molar ratio of 0.01 in photo-assisted Fenton process.
exhibited good accordance to the earlier reports. Hayashi et al.32 showed that [OH˙] in FO typically varies in the range from 1 to 0.01 μM. The average OH˙ generation and CIP degradation rates were found as 3.83 × 10−8 and 2.97 × 10−8 M s−1, respectively. Maezono et al.33 reported a close value of the average OH˙ formation rate as 9.3 × 10−10 M s−1 for the oxidation of Orange II with 60 mg L−1 at pH 3 and Fe2+/H2O2 molar ratio of 0.01 in photo-assisted Fenton process.