
Facile synthesis of submicron-scale layered double hydroxides and their direct decarbonation
Abstract
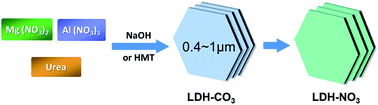
We present a facile synthesis of layered double hydroxides (LDHs) with submicrometer-sized platelets in lateral dimension, and their subsequent direct decarbonation. First, LDH–carbonate phase (LDH_CO3) with submicrometer-sizes in lateral dimension were controlled synthesized by a facile accelerated urea hydrolysis method. Then, using HNO3–NaNO3 mixed solution, the obtained LDHs with carbonate anions in the interlayer were directly decarbonated to their nitrate form (LDH_NO3). These results are important for the synthesis of novel nanomaterials with controlled morphology and will benefit their applications.
 Please wait while we load your content...
Please wait while we load your content...

