Synthesis, structural properties, and pharmacological evaluation of 2-(acylamino)thiophene-3-carboxamides and analogues thereof†
Claudia
Mugnaini
a,
Valentina
Pedani
a,
Daniela
Giunta
b,
Barbara
Sechi
b,
Maurizio
Solinas
b,
Alberto
Casti
c,
Maria Paola
Castelli
c,
Gianluca
Giorgi
a and
Federico
Corelli
*a
aDipartimento di Biotecnologie, Chimica e Farmacia, Università degli Studi di Siena, Via Aldo Moro 2, 53100 Siena, Italy. E-mail: federico.corelli@unisi.it; Fax: +39 0577 234333; Tel: +39 0577 234308
bIstituto di Chimica Biomolecolare, Consiglio Nazionale delle Ricerche, Trav. La Crucca 3 – 07100 Li Punti, Sassari, Italy
cDipartimento di Scienze Biomediche, Divisione di Neuroscienze e Farmacologia Clinica, Cittadella Universitaria, S.S. 554, 09042 Monserrato, Cagliari, Italy
First published on 19th November 2013
Abstract
We have previously reported the synthesis and the pharmacological characterization of a family of methyl 2-(acylamino)thiophene-3-carboxylates as GABAB positive allosteric modulators active both in vitro and in vivo. In the present work, we describe the synthesis of new compounds based on the bioisosteric replacement of the ester moiety with amido or heterocyclic groups as well as of thieno[2,3-d]pyrimidine derivatives as rigid analogues thereof. 4H-Thieno[2,3-d][1,3]oxazin-4-ones were used as synthetic intermediates for the preparation of some of these compounds. The structures of oxazinones 11b, 16, and 17 were assigned by X-ray crystallographic studies, which definitely ruled out the isomeric beta-lactam structure previously hypothesized for these compounds. None of the new molecules exhibited significant activity at the GABAB receptor, either as allosteric or orthosteric ligands.
Introduction
In recent years, remarkable progress has been made in the discovery, chemical optimization and pharmacological understanding of allosteric modulators of G protein-coupled receptors (GPCRs).1–8 An allosteric modulator is a small molecule that binds at a topographically distinct allosteric site of a given receptor and either potentiates or inhibits the binding and/or signaling of an orthosteric ligand. Besides the clinical success of benzodiazepines, the first allosteric modulator drugs which potentiate the effect of the neurotransmitter γ-aminobutyric acid (GABA) at the ionotropic GABAA receptor, the recent development of cinacalcet,9 a positive allosteric modulator (PAM) of the calcium-sensing receptor, and maraviroc,10 a negative allosteric modulator (NAM) of the chemokine receptor CCR5, demonstrated the clinical potential of GPCRs allosteric modulators. In addition to cinacalcet, several highly selective PAMs have been identified for family C GPCRs including calcium-sensing receptors, metabotropic glutamate (mGlu) receptors,1 and GABAB receptors.11–14The GABAB receptor is implicated in different CNS disorders and dysfunctions such as epilepsy, schizophrenia and cognitive impairments, anxiety and depression, pain, drug addiction, and spasticity associated with multiple sclerosis.5,15–19 The search for selective GABAB receptor allosteric potentiators could in principle provide drug candidates for the treatment of a series of diseases and syndromes, including anxiety, osteoarthritic pain, chronic nociceptive pain, alcohol, nicotine and cocaine dependence, and gastro-esophageal reflux disease.20 Based on their mode of action, these novel therapeutic agents are expected to be devoid of drawbacks that characterize full agonists, such as muscle-relaxant effects, hypothermia, central and gastrointestinal side effects, receptor desensitization and tolerance.1,21
Within a research programme aimed at investigating the anti-addiction, particularly anti-alcohol, properties of GABAB receptor PAMs,22 we recently reported the design, synthesis and pharmacological evaluation of a set of 2-(acylamino)thiophene derivatives characterized by good pharmacological, pharmacokinetic and toxicological profiles.23,24 Compounds 1–3 (Fig. 1) were found to be effective in vivo, potentiating baclofen-induced sedation/hypnosis in DBA mice, when administered either intraperitoneally or intragastrically. Although displaying a lower potency in vitro than the reference compound GS39783,12 compounds 1–3 exhibited a higher efficacy in vivo: combination of these compounds with a per se non-sedative dose of baclofen resulted in shorter onset and longer duration of the loss of righting reflex in mice.24
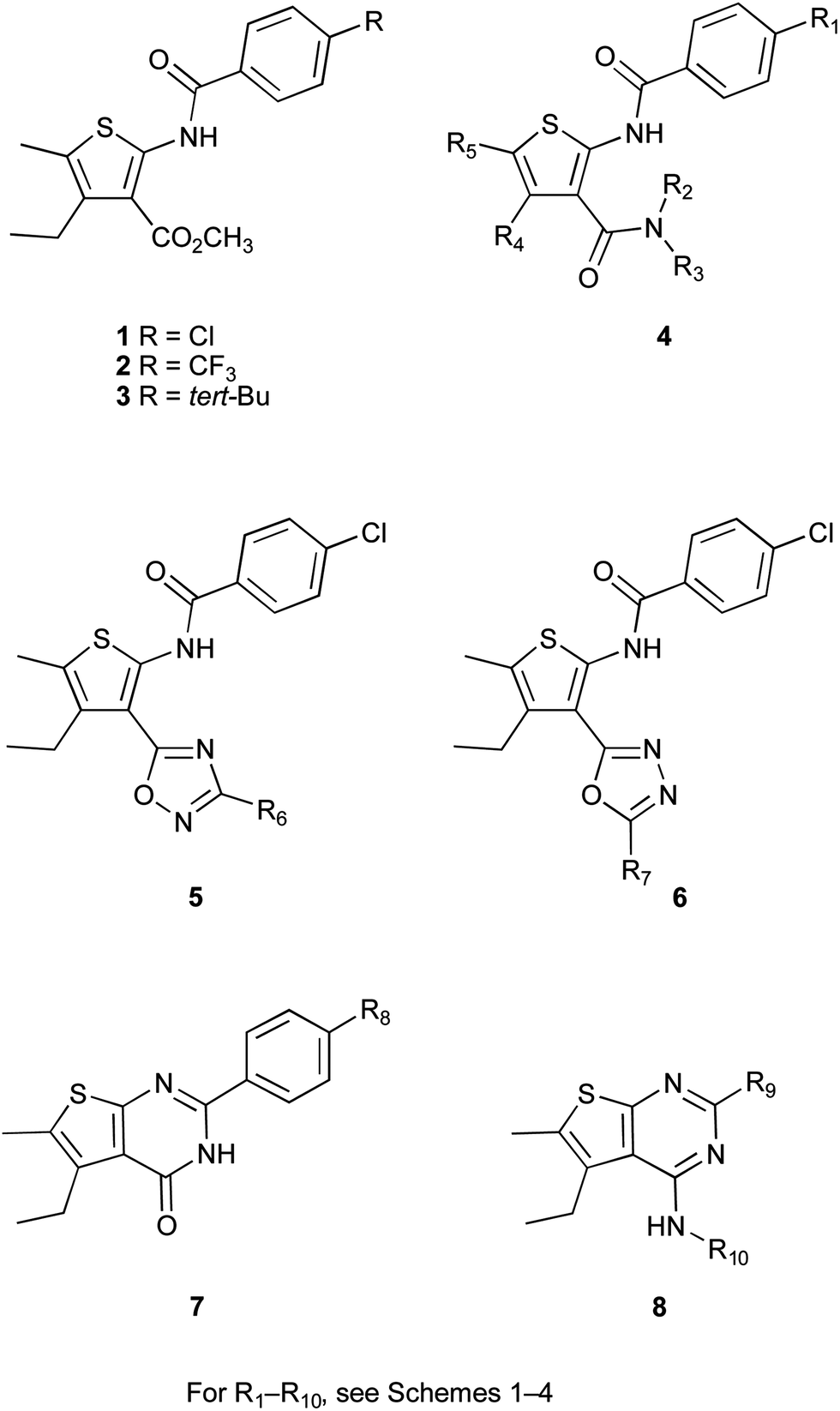 | ||
| Fig. 1 Structure of the lead compounds 1–3 and new analogues 4–8. | ||
These results stimulated our interest in further investigating structure–activity relationship (SAR) of 2-(acylamino)thiophene derivatives. We previously demonstrated that the replacement of the methyl group at the ester moiety by more lipophilic alkyl chains did not improve, but rather decreased, the in vitro activity. Moreover, the hydrolysis of the methyl ester to the corresponding carboxylic acid or the replacement of the ester function by a cyano group led to totally inactive compounds. In a further investigation of SAR, herein we report our efforts aimed at synthesizing new analogues of compounds 1–3 (Fig. 1) where the ester group is replaced by amides (compounds 4) or bioisosteric heterocyclic moieties as 1,2,4-oxadiazole and 1,3,4-oxadiazole rings (compounds 5 and 6).25 Furthermore, thieno[2,3-d]pyrimidines 7 and 8 were also prepared as conformationally restricted analogues of diamides 4.
Results and discussion
Chemistry
The synthesis of compounds 4a–h was performed as highlighted in Scheme 1. Esters 9a–c24 were hydrolyzed to the corresponding carboxylic acids 10a–c.24 On treatment with O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) in DMF, acids 10a–c underwent intramolecular cyclization reaction to yield the oxazinone derivatives 11a–c, which smoothly reacted with primary or secondary amines to provide amides 4a–h in 43–98% yield. Direct reaction between acids 10 and amines in the presence of HBTU or other coupling reagents consistently gave mixtures of amides 4 and oxazinones 11. Therefore, the two-step procedure entailing the isolation of oxazinones was preferred.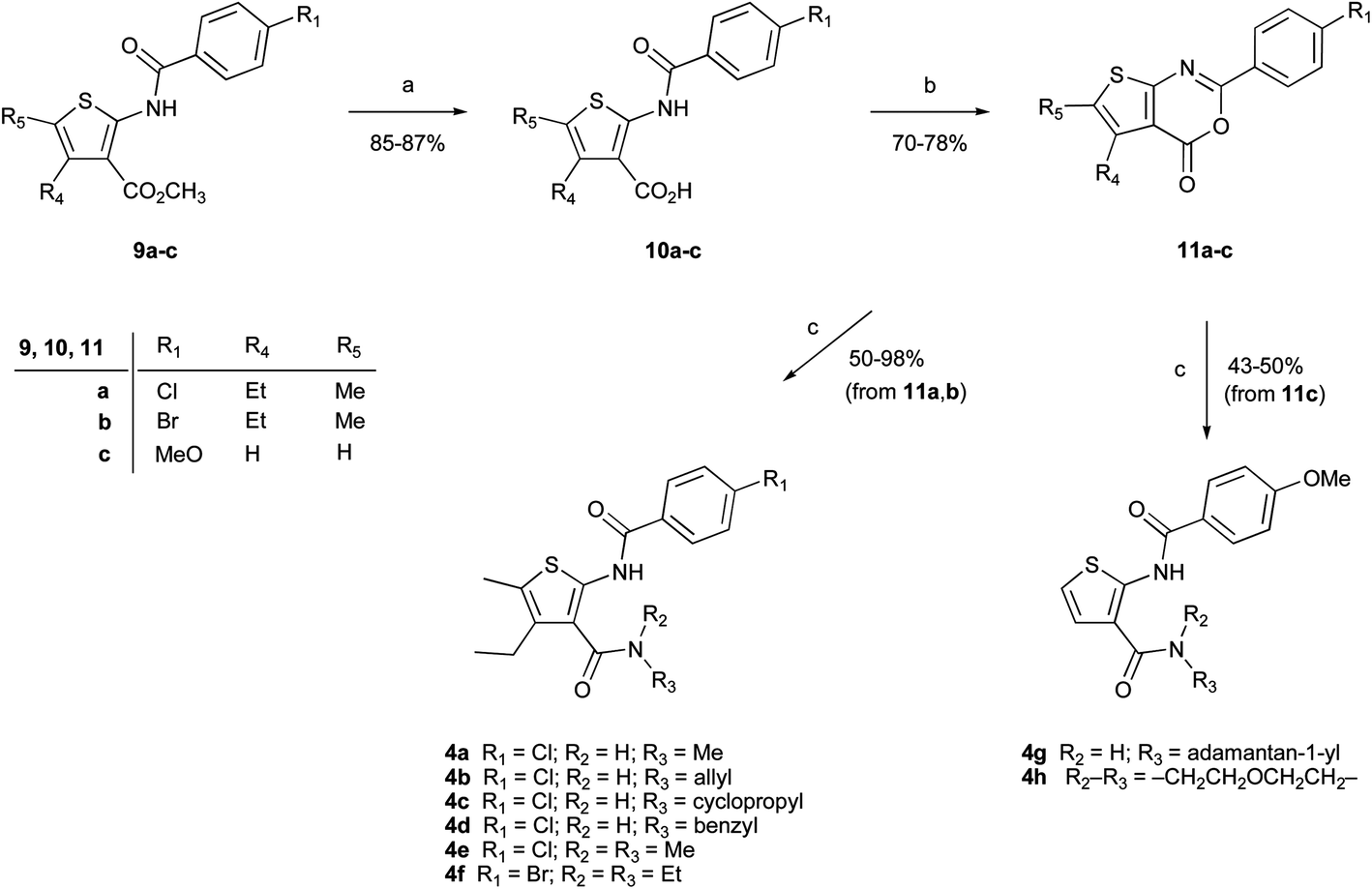 | ||
| Scheme 1 Synthesis of amides 4a–h. (a) NaOH, MeOH/water, 80 °C, 3 h; (b) HBTU, DIPEA, dry DMF, rt, 1 h; (c) appropriate amine, dry DMF, 100 °C, 2.5 h. | ||
Oxazinone 11a was also employed as starting material for the preparation of 1,2,4-oxadiazole derivatives 5a,b (Scheme 2). Benzenecarboximidamide or 1-adamantanecarboximidamide, in turn prepared from the corresponding nitriles according to the procedure recently described,26 were heated with 11a in the presence of potassium tert-butoxide as the base leading to the expected compounds 5a,b, although in modest yield. It is assumed that oxazinone 11a is first converted into intermediate O-acylamidoximes, which subsequently undergo cyclodehydration to the 1,2,4-oxadiazole derivatives 5a,b.27 Compounds 6a,b, bearing the isomeric 1,3,4-oxadiazole moiety, were synthesized from ester 9a in a straightforward manner as detailed in Scheme 3. Hydrazide 12, obtained by treating 9a with an excess of hydrazine hydrate, was reacted with acetyl or benzoyl chloride in refluxing 1,4-dioxane without added base. The intermediate N-acylated hydrazides were converted into the final compounds 6a,b by intramolecular acid-catalyzed dehydration.
 | ||
| Scheme 2 Synthesis of 1,2,4-oxadiazole-5-yl derivatives 5a,b. (a) benzenecarboximidamide or 1-adamantanecarboximidamide, tert-BuOK, dry DMF, reflux, 12 h. | ||
 | ||
| Scheme 3 Synthesis of 1,3,4-oxadiazole-5-yl derivatives 6a,b. (a) Hydrazine hydrate, MeOH, reflux, 20 h; (b) acetyl chloride or benzoyl chloride, dry 1,4-dioxane, 100 °C, 12 h. | ||
Scheme 4 illustrates the preparation of thieno[2,3-d]pyrimidinone and thieno[2,3-d]pyrimidine derivatives 7 and 8. 2-Amino-4-ethyl-5-methythiophene-3-carbonitrile 13 was synthesized by Gewald reaction28 from 3-pentanone and malononitrile, using a modification of the method reported by Barnes et al. employing sodium bicarbonate as a base.29 Conversion of 13 into thieno[2,3-d]pyrimidinone 7a was accomplished by reaction with formic acid in the presence of sulfuric acid as a catalyst under microwave irradiation, according to a protocol recently described.30 Subsequent chlorination of 7a with phosphoryl chloride using microwave heating followed by reaction with cyclohexylamine provided N-cyclohexyl-5-ethyl-6-methylthieno[2,3-d]pyrimidin-4-amine (8a) in 83% yield. The insertion of a cyclohexyl substituent in 8a (as well as in 8f and 8g, vide infra) was suggested by the good pharmacological profile displayed by the cyclohexyl derivative COR628 previously described.23
 | ||
| Scheme 4 Synthesis of thieno[2,3-d]pyrimidinones 7a–d and thieno[2,3-d]pyrimidines 8a–g. (a) Malononitrile, S8, NaHCO3, THF/water, 45 °C, 12 h; (b) HCO2H, H2SO4 (cat.), MW, 90 °C, 10 min; (c) POCl3, MW, 95 °C, 20 min; (d) cyclohexaneamine, EtOH, MW, 150 °C, 30 min; (e) appropriate para-substituted benzoyl chloride, acetonitrile, 78 °C, 3 h; (f) HCO2H, 80 °C, 24 h; (g) appropriate benzonitrile, tert-BuOK, 2-propanol, MW, 100 °C, 30 min; (h) acetic anhydride, 60 °C, 4 h; (i) NaH, cyclohexanecarbonyl chloride, THF, 50 °C, 18 h. | ||
The preparation of amides 14a–c entailed the reaction of 13 with the appropriate para-substituted benzoyl chlorides. We have already shown that the outcome of this apparently ordinary N-acylation reaction is strongly dependent on the experimental conditions used, as considerable amounts of the N,N-diacylated products are formed, particularly in the presence of bases.24 On the contrary, refluxing an acetonitrile solution of 13 in the presence of an equimolar amount of the appropriate para-substituted benzoyl chloride yielded crystalline amides 14a–c upon cooling of the reaction mixture (70–85% yield).
Thieno[2,3-d]pyrimidinones 7b–d were obtained by heating 14a–c with excess formic acid for 24 h (40–68% yield). Aminothieno[2,3-d]pyrimidines 8b–d were obtained by a slight modification of the method described by Chen et al.31 The 2-aminothiophene-3-carbonitrile derivative 13 was directly transformed into 2-substituted 4-aminothieno[2,3-d]pyrimidines 8b–d by reaction with 4-substituted benzonitrile and potassium tert-butoxide under microwave heating in 55–91% yield. It is known that the amino group of compounds such as 8b–d is poorly reactive and only few examples of its functionalization have been reported in the literature.29,31–33 For the synthesis of 8e we initially followed the procedure described by Chen et al. and refluxed 8b in acetic anhydride for 6 hours to obtain the diacylated product. In our hands, after 1.5 hours at 90 °C the substrate was completely converted to 38% of monoacetylated product and 62% of the diacetylated one. Interestingly, we found out that, despite a drop of conversion (85%), the desired compound 8e could be obtained as single reaction product after 4 hours at 60 °C.
In an initial attempt to prepare compounds 8f,g, we tried the procedure reported by Rosowsky et al.32 for similar compounds. However, when 8d and cyclohexanecarbonyl chloride were heated at 40 °C for 24 h in dichloromethane, using triethylamine as a base, we did not observe any conversion of the starting material. Conversely, using a stronger base, such as sodium hydride in THF, after 18 h at 50 °C the amino derivatives 8c and 8d could be transformed into the corresponding cyclohexanecarboxamides 8f and 8g in excellent yields (82% and 99%, respectively).
X-Ray crystallography
A number of papers reported the synthesis and reactivity of hetero-1,3-oxazinones, in particular 4H-isothiazolo[5,4-d][1,3]oxazin-4-ones which have been used as intermediates for the preparation of the antiviral agent denotivir and related compounds.34 The first synthesis of a compound of this type, i.e. 6-phenyl-3-methyl-4H-isothiazolo[5,4-d][1,3]oxazin-4-one (15a, Fig. 2), containing the considered ring system, was described in 1969, but at that time the interpretation of its structure was incorrect, as it was presented as 5-benzoyl-3-methylisothiazolo[5,4-b]azetin-4-one (15b, Fig. 2).35 In 2006, Regiec and coworkers reported the synthesis of 15a, the structure of which was assigned on the basis of IR spectrum.34 In spite of the wide synthetic exploitation of hetero-1,3-oxazinones, to our knowledge no definitive structural assignment based on X-ray crystallography has been reported so far, particularly with regard to 4H-thieno[2,3-d][1,3]oxazin-4-ones 11 which were used as synthetic intermediates in our work. Accordingly, we decided to analyze the structure of the representative thieno[2,3-d][1,3]oxazin-4-one derivative 11b through X-ray diffraction studies and its crystal structure is reported in Fig. 3. | ||
| Fig. 2 Oxazinone structure 15avs. beta-lactam structure 15b and other heterocondensed oxazinones characterized by X-ray crystallography. | ||
 | ||
| Fig. 3 X-ray crystal structure of compound 11b. Ellipsoids enclose 50% probability. | ||
In the thienoxazine system, the O(3)–C(2) bond length (1.365(3) Å) is shorter than O(3)–C(4) (1.408(3) Å) and the bond angles at C(2) and C(4) are distorted from their ideal values of 120°. The largest deviation is shown by the exocyclic O(4)–C(4)–C(5) bond angle with a value of 130.1(2)°. Such observations can be explained as a result of a concerted electronic effects of adjacent substituents.36 The thienoxazinone system is essentially planar, in agreement with the only one crystal structure of a thieno[2,3-d][1,3]oxazin-4-one derivative37 reported in the Cambridge Structural Database (CSD) ver. 5.34, and with experimental data of bioisosteric benzoxazinones.36,38 The largest deviation (−0.016(1) Å) from the thienoxazine least squares plane is shown by O(3). The phenyl ring is almost co-planar with the thienoxazinone system forming a dihedral angle of 2.68(9)°. This conformation causes the existence of two intramolecular non-conventional hydrogen bonding interactions between C(6′)-H(6′)⋯O(3) and C(2′)-H(2′)⋯N(1) with H⋯O/N distances of 2.39(1) and 2.55(1) Å, respectively.
The structural investigation was extended to 1,3-oxazinones condensed with different heterocyclic rings (compounds 16 and 17, Fig. 2). Therefore, following the procedures adopted for the preparation of oxazinones 11a–c, methyl 3-amino-1H-pyrrole-2-carboxylate 1839 and the commercially available ethyl 5-amino-1-methyl-1H-pyrazole-4-carboxylate 19 (Scheme 5) were acylated to the amides 20 and 21, respectively. Subsequent hydrolysis to the corresponding acids, followed by cyclodehydration, afforded the expected oxazinones 16 and 17.
 | ||
| Scheme 5 Synthesis of oxazinone derivatives 16 and 17. (a) Benzoyl or p-chlorobenzoyl chloride, acetonitrile, 78 °C, 3 h; (b) i NaOH, MeOH–water, 80 °C, 3 h; ii HBTU, DIPEA, dry DMF, rt, 1 h. | ||
The crystal structures of oxazinones 16·CH3OH and 17 (Fig. 4) are of interest because no structure of pyrazolo[3,4-d][1,3]oxazin-4(1H)-one or pyrrolo[3,2-d][1,3]oxazin-4(5H)-one derivatives is present in the CSD.
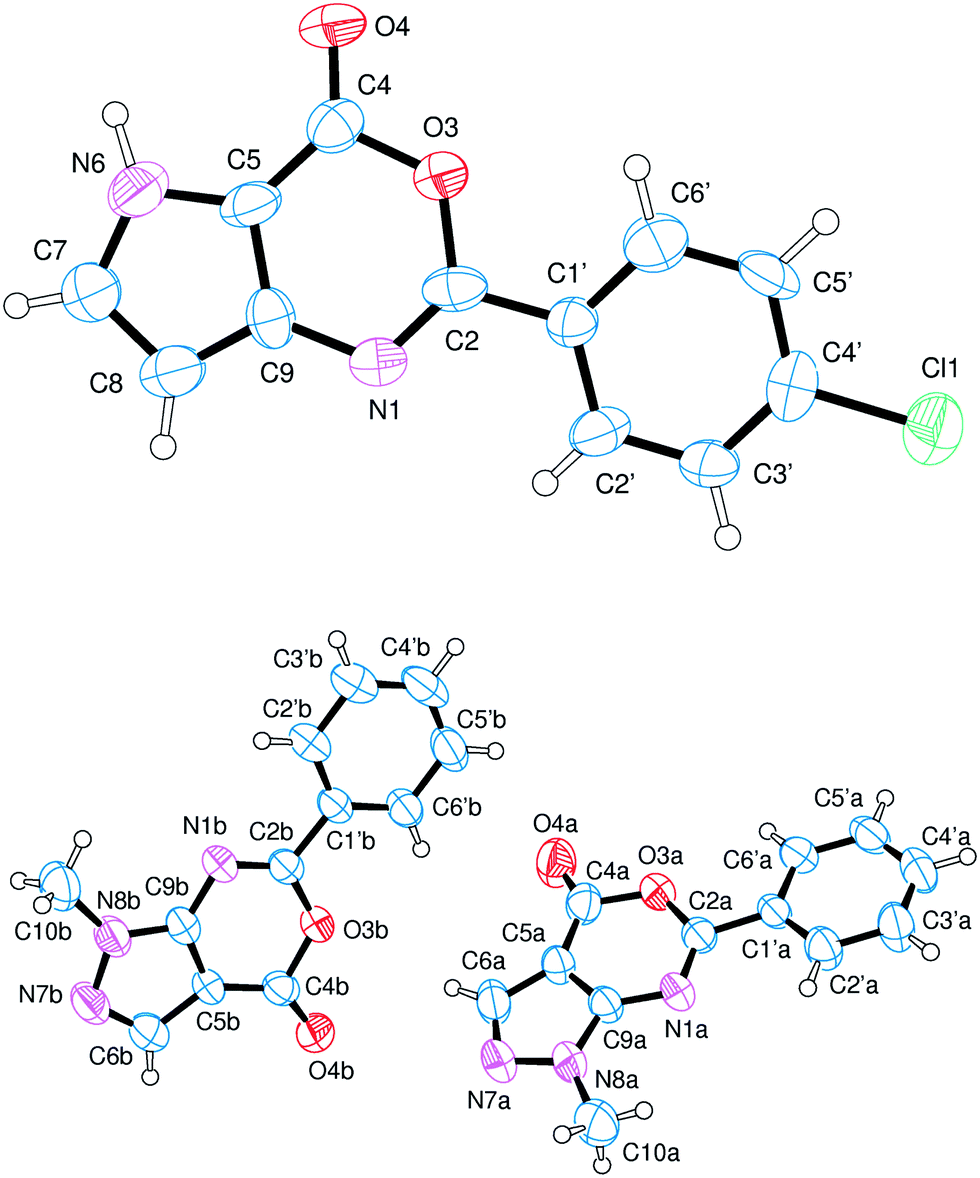 | ||
| Fig. 4 X-ray crystal structure of compounds 16·CH3OH (top; methanol moiety omitted for clarity) and 17 (bottom). Ellipsoids enclose 50% probability. | ||
Likely to the thienoxazine system, also in the two oxazinone derivatives the O(3)–C(2) bond length is shorter than O(3)–C(4) of 0.039(7), 0.065(2) and 0.048(2) Å in 16·CH3OH and in the two molecules of 17, respectively. Further, the bond angles at C(2) and C(4) are distorted from their ideal values of 120°. In particular the bond angle O(4)–C(4)–C(5) has values of 130.9(6), 132.6(2) and 132.2(2)° in 16·CH3OH and in the two molecules of 17, respectively.
In the 29 entries of oxazinone derivatives found in CSD, the value of this bond angle ranges from 126.1°, found in 2-(1-amino-1-phenylprop-1-en-2-yl)-5-methyl-4-phenyl-6H-[1,3]-oxazin-6-one,40 to 132.9° found in 5-amino-3-β-D-ribofuranosyl-3H-imidazo[4,5-d][1,3]-oxazin-7-one hemihydrate.41
The pyrazolo- and the pyrrolo-oxazine systems are almost planar with the largest deviations shown by C(8) (0.0022(6) Å) in 16·CH3OH and by N(7A) (0.016(2) Å) and O(3B) (0.014(1) Å) in the two molecules of 17.
The structure of 16·CH3OH is stabilized by intermolecular hydrogen bonding N(6)–H(6)(x,y,z)⋯O(4)(−x, 3−y, −z) interactions with a H⋯O distance equal to 1.91(6) Å. Stacking interactions are also present between molecules in (x,y,z) and (x,y−1,z) with an interplanar distance of 3.8(1) Å (Fig. 5).
 | ||
| Fig. 5 Hydrogen bonding and stacking interactions in 16·CH3OH (molecules of methanol are omitted for clarity). | ||
The results of these crystallographic studies definitely confirm that intramolecular cyclization of heterocyclic analogs of anthranilic acid amides, under the experimental conditions adopted by us and other researchers, leads to condensed 1,3-oxazinones instead of the corresponding beta-lactams. This is most likely due to the low stability of the condensed Δ3-azetinone structure (e.g.15b).42 To our knowledge, only few examples of N-tert-alkylbenzoazetinones obtained by ring opening/recyclization of anthranilium ions, proved to be sufficiently stable to be isolated and characterized.43
In vitro pharmacology
The functional characterization of the novel compounds 4–8 was performed using GTPγS binding assay, a well validated functional assay for GPCRs, using membranes from rat brain cortex.23,24 To assay for GABAB PAM activity, the compounds were co-applied with the endogenous ligand GABA. In the presence of 10 μM GABA, the test compounds at a concentration up to 25 μM did not potentiate [35S]GTPγS stimulation induced by GABA alone by more than 4%, thereby resulting substantially inactive.However disappointing these results may be, they are not totally surprising. Early efforts aimed at developing SAR for GPCRs allosteric modulators revealed that the phenomenon known as “flat SAR” appears considerably more widespread than with orthosteric ligands,1,44 introducing significant challenges in the hit-to-lead stage of drug discovery process.45 Accordingly, even slight modifications of the hit structure may result into inactive compounds or point toward very shallow and narrow SAR patterns. Flat SAR is frequently observed with PAMs,46 and the compounds described herein do not appear to be an exception. As a result, further studies should be undertaken to develop a consistent and comprehensive SAR model, which may act as a guide in the rational design of novel analogues and the hit-to-lead optimization process.
Conclusions
Within a research program aimed at discovering new positive allosteric modulators of the GABAB receptor, we presented here the synthesis of a series of amide analogues, oxadiazole derivatives as well as conformationally rigid thieno[2,3-d]pyrimidines and thieno[2,3-d]pyrimidones starting from the same class of 2-aminothiophene-3-carboxy or 3-carbonitrile derivatives. In particular we found out that a series of oxazinones can be synthesized in good yield and successfully used as intermediates for the preparation of the amide analogues through nucleophilic opening by the appropriate amines. Interestingly, among these intermediates, the structure of 2-(4-bromophenyl)-5-ethyl-6-methyl-4H-thieno[2,3-d][1,3]oxazin-4-one (11b) was resolved by X-ray crystallography, thereby ruling out definitely the isomeric beta-lactam structure that had been previously hypothesized for this compound. When subjected to GTPγS binding assay, none of the new compounds revealed significant GABAB receptor modulatory activity.In spite of these pharmacological results, we have provided valid synthetic strategies for the preparation of compounds 4–8 with good to excellent chemical yields. Every new information in the still largely unexplored field of GABAB PAMs may be useful to stimulate further investigations and speed up the drug discovery process.
Experimental section
General information
Reagents were purchased from commercial suppliers and used without further purification. All reactions were run under a positive pressure of dry N2 unless otherwise specified. Merck silica gel 60 was used for flash chromatography (23–400 mesh). IR spectra were recorded on a Perkin-Elmer BX FT-IR system (CHCl3 solution) or on KBr disks using a Thermo Nicolet Avatar 330 FT-IR. 1H NMR and 13C NMR were recorded at 400 and 100 MHz, respectively, on a Bruker Advance DPX400 or on a Varian Mercury instrument. Chemical shifts are reported in ppm and multiplicity is specified as follow: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, bs = broad signal. Mass spectral (MS) data were obtained using Agilent 1100 LC/MSD VL system (G1946C). Mass spectra were acquired either in positive or in negative mode scanning and data are reported as m/z. Melting points were determined on either a Gallenkamp apparatus or a Büchi B-540 instrument and are uncorrected. Microwave irradiations were conducted using a CEM Discover Synthesis Unit (CEM Corp., Matthews, NC) equipped with a 300 W power source. Purity of tested compounds is ≥95% as determined by elemental analysis. Elemental analyses were performed on a Perkin-Elmer PE 2004 elemental analyzer and the data for C, H, and N are within 0.4% of the theoretical values.General procedure for the synthesis of oxazinones 11a–c
To a solution of the appropriate carboxylic acid 10a–c (1 mmol) in dry DMF (5 mL) were added DIPEA (0.4 mL, 2.35 mmol) and HBTU (380 mg, 1 mmol). The reaction mixture was stirred at room temperature under N2 atmosphere for 1 h. The mixture was poured into water and extracted in dichloromethane. The organic layer was washed with brine, dried over anhydrous sodium sulfate and evaporated to dryness. The crude product was purified by flash column chromatography using dichloromethane–petroleum ether (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as eluent.
:1) as eluent.
General procedure for the synthesis of diamides 4a–h
The appropriate amine (1.2 mmol) was added to a solution of oxazinone 11a–c (1 mmol) in dry DMF (7 mL) and the reaction mixture was stirred at 100 °C under N2 atmosphere for 2.5 h. Then the mixture was poured into water and extracted with dichloromethane. The organic layer was washed with brine, dried over anhydrous sodium sulfate and evaporated to dryness. The crude product was purified by flash column chromatography using ethyl acetate–petroleum ether (1:1) as eluent or triturated from diethyl ether to give pure diamide derivates 4a–h.
General procedure for the synthesis of the 1,2,4-oxadiazole derivates 5a,b
To a solution of 11a (305 mg, 1 mmol) in dry DMF (7.5 mL), were added potassium tert-butoxide (112 mg, 1 mmol) and the appropriate amidoxime (1 mmol). The reaction mixture was stirred under reflux in a N2 atmosphere for 12 h. The mixture was poured into water and extracted with dichloromethane. The organic layer was washed with brine, dried over anhydrous sodium sulfate and evaporated to dryness. The crude product was purified by flash column chromatography using dichloromethane as eluent to give the title compounds.General procedure for the synthesis of 1,3,4-oxadiazole derivatives 6a,b
To a solution of compound 9a (337 mg, 1 mmol) in dry 1,4-dioxane (18 mL), the appropriate acyl chloride (1 mmol) was added. The reaction mixture was stirred at 70 °C for 12 h, then at 100 °C for 4 h. The mixture was concentrated under reduced pressure and the residue was dissolved in ethyl acetate and washed with brine. The organic layer was dried over anhydrous sodium sulfate and evaporated to dryness. The crude product was purified by flash column chromatography using ethyl acetate–petroleum ether (1:1) as eluent to give the title compound.
:2) afforded 13 in 80% yield as a white solid, mp 104–106 °C.47
:1), then methanol, to provide the title compound 7a in 95% yield. Mp 178–180 °C. 1H NMR (CDCl3): δ 8.05 (s, 1H), 2.97 (q, J = 7.4 Hz, 2H), 2.44 (s, 3H), 1.20 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3): δ 163.3, 159.8, 142.9, 136.1, 131.1, 123.5, 20.8, 15.3, 12.9. IR (KBr): ν 3163, 1662, 1577 cm−1. MS (ESI): m/z 195 [M + H]+ (100). Anal. calcd for C9H10N2OS: C, 55.65; H, 5.19; N, 14.42. Found: C, 55.81; H, 5.11; N, 14.27%.
General procedure for the synthesis of benzamides 14a–c, 20, 21
The appropriate para-substituted benzoyl chloride (3 mmol) was added to a solution of the amino derivative 13, 18, 19 (3 mmol) in acetonitrile (10 mL). The mixture was stirred at 78 °C for 3 h, then cooled to room temperature to allow product precipitation. The product was collected by filtration and washed with acetonitrile to afford pure 14a–c, 20, 21.General procedure for the synthesis of 5-ethyl-6-methyl-2-(aryl)thieno[2,3-d]pyrimidin-4(3H)-ones (7b–d)
The appropriate benzamide 14a–c (0.3 mmol) and formic acid (10 mL) were stirred in a 50 mL round bottom flask for 24 hours at 80 °C. After cooling to room temperature the reaction mixture was poured onto ice cold water (50 mL) and extracted with dichloromethane. The organic phase was dried over sodium sulfate, filtered and the solvent removed under vacuum. Purification of the residue by flash chromatography or crystallization from diethyl ether afforded the desired product.:3) as eluent. Mp 236–238 °C. 1H NMR (CDCl3): δ 13.2 (bs, 1H), 8.12 (d, J = 8.2 Hz, 2H), 7.77 (d, J = 8.2 Hz, 2H), 2.76 (q, J = 7.8 Hz, 2H), 2.36 (s, 3H), 1.26 (t, J = 7.8 Hz, 3H). 13C NMR (CDCl3): δ 168.5, 162.1, 145.9, 135.9, 133.8 (q, 2J[CF] = 33.9 Hz), 127.9, 125.8 (q, 3J[CF] = 3.9 Hz), 125.1, 123.6 (q, 1J[CF] = 272 Hz), 114.3, 21.1, 14.6, 12.3. IR (KBr): ν 3469, 1674 cm−1. MS (ESI): m/z 339 [M + H]+, 377 [M + K]+ (100). Anal. calcd for C16H13F3N2OS: C, 56.80; H, 3.87; N, 8.28. Found: C, 57.10; H, 3.89; N, 8.16%.
:1) yielded 8a in 83% yield as a white solid, mp 92–93 °C. 1H NMR (CDCl3): δ 8.40 (s, 1H), 5.40 (bs, 1H), 4.26 (m, 1H), 2.81 (q, J = 7.8 Hz, 2H), 2.44 (s, 3H), 2.11 (m, 2H), 1.75 (m, 2H), 1.68 (m, 1H), 1.51 (m, 2H), 1.33 (m, 3H) overlapped with 1.28 (t, J = 7.8 Hz, 3H). 13C NMR (CDCl3): δ 164.0, 155.9, 152.2, 129.9, 129.7, 116.1, 48.9, 33.0, 25.6, 24.5, 21.5, 14.9, 13.0. IR (KBr): ν 3420, 1565 cm−1. MS (ESI): m/z 276 [M + H]+ (100). Anal. calcd for C15H21N3S: C, 65.41; H, 7.69; N, 15.26. Found: C, 65.65; H, 7.60; N, 15.09%.
General procedure for the synthesis of 2-(aryl)thieno[2,3-d]pyrimidin-4-amine (8b–d)
A mixture of 13 (200 mg, 1.2 mmol), the appropriate aromatic nitrile (1.44 mmol) and potassium tert-butoxide (273 mg, 2.4 mmol) in 2-propanol (3 mL) was subjected to microwave irradiation for 30 min at 100 °C. After cooling, the reaction mixture was poured onto ice and the solid residue was filtered and dissolved in dichloromethane. The organic phase was washed with H2O, dried over sodium sulfate, filtered and evaporated under vacuum. Purification of the residue by flash chromatography yielded the desired products.:3) as eluent. White solid, mp 210–212 °C. 1H NMR (CDCl3): δ 8.54 (d, J = 8.2 Hz, 2H), 7.71 (d, J = 8.2 Hz, 2H), 5.51 (bs, 2H), 2.84 (q, J = 7.7 Hz, 2H), 2.50 (s, 3H), 1.31 (t, J = 7.7 Hz, 3H). 13C NMR (CDCl3): δ 167.4, 157.0, 156.8, 140.9, 131.4 overlapped with 131.4 (q, 2J[CF] = 32.2 Hz), 128.2, 125.2 (q, 3J[CF] = 3.8 Hz), 125.1, 124.2 (q, 1J[CF] = 273 Hz), 115.0, 21.4, 15.2, 13.2. IR (KBr): ν 3494, 1614 cm−1. MS (ESI): m/z 338 [M + H]+ (100). Anal. calcd for C16H14F3N3S: C, 56.96; H, 4.18; N, 12.46. Found: C, 57.19; H, 4.11; N, 12.58%.
:3). Pale yellow solid, mp 185–187 °C. 1H NMR (CDCl3): δ 8.37 (d, J = 8.6 Hz, 2H), 7.43 (d, J = 8.6 Hz, 2H), 5.48 (bs, 2H), 2.83 (q, J = 7.8 Hz, 2H), 2.48 (s, 3H), 1.30 (t, J = 7.8 Hz, 3H). 13C NMR (CDCl3): δ 167.6, 156.2, 136.8, 134.9, 131.8, 130.6, 129.5, 128.9, 114.7, 21.6, 15.3, 13.4. IR (KBr): ν 3482, 1600 cm−1. MS (ESI): m/z 304 [M + H]+ (100), 305 [M + 2H]+. Anal. calcd for C15H14ClN3S: C, 59.30; H, 4.64; N, 13.83. Found: C, 59.53; H, 4.72; N, 13.60%.
:3) as eluent gave pure 8e (870 mg, 85%). White solid, mp 220–221 °C. 1H NMR (DMSO-d6): δ 10.6 (s, 1H), 8.59 (d, J = 8.2 Hz, 2H), 7.89 (d, J = 8.2 Hz, 2H), 2.83 (q, J = 7.4 Hz, 2H), 2.54 (s, 3H), 2.24 (s, 3H), 1.08 (t, J = 7.4 Hz, 3H). 13C NMR (DMSO-d6): δ 170.5, 169.2, 156.0, 151.6, 140.1, 135.1, 131.5, 130.4 (q, 2J[CF] = 31.6 Hz), 128.3, 125.4 (q, 3J[CF] = 4.0 Hz) overlapped with 124.1 (q, 1J[CF] = 272.7 Hz), 123.8, 23.2, 19.8, 14.5, 13.4. IR (KBr): ν 3226, 1700 cm−1. MS (ESI): m/z 380 [M + H]+ (100). Anal. calcd for C18H16F3N3OS: C, 56.98; H, 4.25; N, 11.08. Found: C, 57.21; H, 4.19; N, 10.89%.
General procedure for the synthesis of cyclohexanecarboxamide derivatives 8f,g
A mixture of the appropriate amine 8c,d (0.35 mmol) and 97% NaH (86 mg, 3.5 mmol) were stirred under nitrogen at room temperature in 5 mL of dry THF. After addition of cyclohexanecarbonyl chloride (56 mg, 0.38 mmol) the reaction mixture was stirred at 50 °C for 18 hours. Cold water was carefully added to the reaction mixture cooled in an ice bath. After addition of dichloromethane, the organic phase was separated, dried over anhydrous sodium sulfate, and filtered. Removal of the solvent under vacuum gave a solid residue, which was purified by washing with cold THF to provide the desired products.General procedure for the synthesis of oxazinones 16 and 17
Amidoester 20, 21 were hydrolyzed to the corresponding carboxylic acids following the same procedure described for the preparation of 10a–c. The carboxylic acids were directly converted into the title compounds according to the general procedure for oxazinone synthesis reported above.X-Ray crystallography
Single crystals of 11b, 16·CH3OH and 17 were submitted to X-ray data collections by using a Xcalibur, Sapphire3 (Oxford Diffraction Ltd., U.K.) four-circle diffractometer with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods implemented in SHELXS-97 program.48 The refinements were carried out by full-matrix anisotropic least-squares on F2 for all reflections for non-H atoms by using the SHELXL program.49In vitro binding studies
Tissue preparation and [35S]GTPγS binding assay in rat cortical membranes were carried out as previously described.23,24Acknowledgements
This work was partially supported by grant CRP-25257 to MPC and to MS from Regione Autonoma della Sardegna (L.R. 7 agosto 2007, n. 7).References and notes
- S. Urwyler, Pharmacol. Rev., 2011, 63, 59–126 CrossRef CAS PubMed.
- B. J. Melancon, C. R. Hopkins, M. R. Wood, K. A. Emmitte, C. M. Niswender, A. Christopoulos, P. J. Conn and C. W. Lindsley, J. Med. Chem., 2012, 55, 1445–1464 CrossRef CAS PubMed.
- N. T. Burford, J. Watson, R. Bertekap and A. Alt, Biochem. Pharmacol., 2011, 81, 691–702 CrossRef CAS PubMed.
- P. Keov, P. M. Sexton and A. Christopoulos, Neuropharmacology, 2011, 60, 24–35 CrossRef CAS PubMed.
- M. De Amici, C. Dallanoce, U. Holzgrabe, C. Trankle and K. Mohr, Med. Res. Rev., 2010, 30, 463–549 CAS.
- R. Franco, E. I. Canela, V. Casado and S. Ferre, Expert Opin. Drug Discovery, 2010, 5, 391–403 CrossRef CAS PubMed.
- L. Wang, B. Martin, R. Brenneman, L. M. Luttrell and S. Maudsley, J. Pharmacol. Exp. Ther., 2009, 331, 340–348 CrossRef CAS PubMed.
- P. J. Conn, A. Christopoulos and C. W. Lindsley, Nat. Rev. Drug Discovery, 2009, 8, 41–54 CrossRef CAS PubMed.
- P. E. Harrington and C. Fotsch, Curr. Med. Chem., 2007, 14, 3027–3034 CrossRef CAS.
- P. Dorr, M. Westby, S. Dobbs, P. Griffin, B. Irvine, M. Macartney, J. Mori, G. Rickett, C. Smith-Burchnell, C. Napier, R. Webster, D. Armour, D. Prince, B. Stammen, A. Wood and M. Perros, Antimicrob. Agents Chemother., 2005, 49, 4721–4732 CrossRef CAS PubMed.
- S. Urwyler, J. Mosbacher, K. Lingenhoehl, J. Heid, K. Hofstetter, W. Froestl, B. Bettler and K. Kaupmann, Mol. Pharmacol., 2001, 60, 963–971 CAS.
- S. Urwyler, M. F. Pozza, K. Lingenhoehl, J. Mosbacher, C. Lampert, W. Froestl, M. Koller and K. Kaupmann, J. Pharmacol. Exp. Ther., 2003, 307, 322–330 CrossRef CAS PubMed.
- S. Guery, P. Floersheim, K. Kaupmann and W. Froestl, Bioorg. Med. Chem. Lett., 2007, 17, 6206–6211 CrossRef CAS PubMed.
- P. Malherbe, R. Masciadri, R. D. Norcross, F. Knoflach, C. Kratzeisen, M. T. Zenner, Y. Kolb, A. Marcuz, J. Huwyler, T. Nakagawa, R. H. Porter, A. W. Thomas, J. G. Wettstein, A. J. Sleight, W. Spooren and E. P. Prinssen, Br. J. Pharmacol., 2008, 154, 797–811 CrossRef CAS PubMed.
- B. Bettler, K. Kaupmann, J. Mosbacher and M. Gassmann, Physiol. Rev., 2004, 84, 835–867 CrossRef CAS PubMed.
- C.-M. Vacher and B. Bettler, Curr. Drug Targets: CNS Neurol. Disord., 2003, 2, 248–259 CrossRef CAS.
- J. Ong and D. I. B. Kerr, CNS Drug Rev., 2005, 11, 317–334 CrossRef CAS.
- N. G. Bowery, Curr. Opin. Pharmacol., 2006, 6, 37–43 CrossRef CAS PubMed.
- R. Lujan and F. Ciruela, Curr. Drug Targets, 2012, 13, 129–144 CrossRef CAS.
- W. Froestl, Future Med. Chem., 2011, 3, 163–175 CrossRef CAS PubMed.
- J. P. Pin and L. Prézeau, Curr. Neuropharmacol., 2007, 5, 195–201 CrossRef CAS PubMed.
- P. Maccioni, A. Zaru, B. Loi, C. Lobina, M. A. M. Carai, G. L. Gessa, C. Mugnaini, S. Pasquini, F. Corelli, P. Hyytiä, L. Lumeng and G. Colombo, Alcohol.: Clin. Exp. Res., 2012, 36, 1748–1766 CrossRef CAS PubMed.
- M. P. Castelli, A. Casu, P. Casti, C. Lobina, M. A. M. Carai, G. Colombo, M. Solinas, D. Giunta, C. Mugnaini, S. Pasquini, A. Tafi, S. Brogi, G. L. Gessa and F. Corelli, J. Pharmacol. Exp. Ther., 2012, 340, 529–538 CrossRef CAS PubMed.
- C. Mugnaini, V. Pedani, A. Casu, C. Lobina, A. Casti, P. Maccioni, A. Porcu, D. Giunta, S. Lamponi, M. Solinas, S. Dragoni, M. Valoti, G. Colombo, M. P. Castelli, G. L. Gessa and F. Corelli, J. Med. Chem., 2013, 56, 3620–3635 CrossRef CAS PubMed.
- C. Mugnaini, S. Nocerino, V. Pedani, S. Pasquini, A. Tafi, M. De Chiaro, L. Bellucci, M. Valoti, F. Guida, L. Luongo, S. Dragoni, A. Ligresti, A. Rosenberg, D. Bolognini, M. G. Cascio, R. G. Pertwee, R. Moaddel, S. Maione, V. Di Marzo and F. Corelli, ChemMedChem, 2012, 7, 920–934 CrossRef CAS PubMed.
- G. Xia, X. You, L. Liu, H. Liu, J. Wang, Y. Shi, P. Li, B. Xiong, X. Liu and J. Shen, Eur. J. Med. Chem., 2013, 62, 1–10 CrossRef CAS PubMed.
- J. A. Tucker, T. L. Clayton, C. G. Chidester, M. W. Schulz, L. E. Harrington, S. J. Conrad, Y. Yagi, N. L. Oien, D. Yurek and M. S. Kuo, Bioorg. Med. Chem., 2002, 8, 601–615 CrossRef.
- For a recent review on the Gewald reaction, see: Y. Huang and A. Dömling, Mol. Diversity, 2011, 15, 3–33 CrossRef CAS PubMed.
- D. M. Barnes, A. R. Haight, T. Hameury, M. A. McLaughlin, J. Mei, J. S. Tedrow and J. Dalla Riva Toma, Tetrahedron, 2006, 62, 11311–11319 CrossRef CAS PubMed.
- S. Hesse, E. Perspicace and G. Kirsch, Tetrahedron Lett., 2007, 48, 5261–5264 CrossRef CAS PubMed.
- B. Chen, E. Perspicace, S. Hesse and G. Kirsch, Synthesis, 2010, 2413–2418 CAS.
- A. Rosowsky, A. T. Papoulis and S. F. Queener, J. Med. Chem., 1997, 40, 3694–3699 CrossRef CAS PubMed.
- Y. Dai, Y. Guo, R. R. Frey, Z. Ji, M. L. Curtin, A. A. Ahmed, D. H. Albert, L. Arnold, S. S. Arries, T. Barlozzari, J. L. Bauch, J. J. Bouska, P. F. Bousquet, G. A. Cunha, K. B. Glaser, J. Guo, J. Li, P. A. Marcotte, K. C. Marsh, M. D. Moskey, L. J. Pease, K. D. Stewart, V. S. Stoll, P. Tapang, N. Wishart, S. K. Davidsen and M. R. Michaelides, J. Med. Chem., 2005, 48, 6066–6083 CrossRef CAS PubMed.
- A. Regiec, Z. Machon, R. Miedzybrodzki and S. Szymaniec, Arch. Pharm. Chem. Life Sci., 2006, 339, 401–413 CrossRef CAS PubMed.
- Z. Machon, Diss. Pharm. Pharmacol., 1969, XXI, 325–335 Search PubMed , Chem. Abstr. 1970, 72, 43552k.
- A. Yu. Kovalevsky and I. I. Ponomarev, Acta Crystallogr., Sect. A: Found. Crystallogr., 2000, C56, 260–262 CAS.
- M. Pietsch, M. Nieger and M. Gutschow, Acta Crystallogr., Sect. A: Found. Crystallogr., 2007, C63, o147–o151 Search PubMed.
- (a) J. D. Crane and E. Rogerson, Acta Crystallogr., Sect. A: Found. Crystallogr., 2004, E60, o669–o670 Search PubMed; (b) M. Gütschow, U. Neumann, J. Sieler and K. Eger, Pharm. Acta Helv., 1998, 73, 95–103 CrossRef; (c) A. Yu. Kovalevsky, I. I. Ponomarev and M. A. Baranova, Acta Crystallogr., Sect. A: Found. Crystallogr., 2000, C56, e408–e409 CAS; (d) B. N. Yadav, S. Prasad and S. M. Prasad, Acta Crystallogr., Sect. A: Found. Crystallogr., 2002, E58, o1111–o1112 Search PubMed.
- R. H. Fourneaux and P. C. Tyler, J. Org. Chem., 1999, 64, 8411–8412 CrossRef.
- Y. S. Chun, J. H. Kim, S. Y. Choi, Y. O. Ko and S.-g. Lee, Org. Lett., 2012, 14, 6358–6361 CrossRef CAS PubMed.
- H. Nakamura, N. Yagisawa, N. Shimada, T. Takita, H. Umezawa and Y. Iitaka, J. Antibiot., 1981, 34, 1219–1221 CrossRef CAS.
- S.-J. Chiu and C.-H. Chou, Tetrahedron Lett., 1999, 40, 9271–9272 CrossRef CAS.
- R. A. Olofson, R. K. Vander Meer, D. H. Hoskin, M. Y. Bernheim, S. Stournas and D. S. Morrison, J. Org. Chem., 1984, 49, 3367–3372 CrossRef CAS.
- P. J. Conn, S. D. Kuduk and D. Doller, in Annual Reports in Medicinal Chemistry, ed. M. C. Desai, Academic Press, San Diego, 2012, vol. 47, pp. 441–457 Search PubMed.
- R. Williams, C. M. Niswender, Q. Luo, U. Le, P. J. Conn and C. W. Lindsley, Bioorg. Med. Chem. Lett., 2009, 19, 962–966 CrossRef CAS PubMed.
- C. R. Hopkins, C. W. Lindslay and C. M. Niswender, Future Med. Chem., 2009, 1, 501–513 CrossRef CAS PubMed.
- Different physico-chemical properties have been reported for this compound in a number of publications, see for example: (a) K. Gewald, Chem. Ber., 1965, 98, 3571–3577 CrossRef CAS; (b) T. Charvát, M. Potácek and J. Marek, Monatsh. Chem., 1995, 126, 333–340 CrossRef; (c) X.-G. Huang, J. Liu, J. Ren, T. Wang, W. Chen and B.-B. Zeng, Tetrahedron, 2011, 67, 6202–6205 CrossRef CAS PubMed; (d) R. Tayebee, F. Javadi and G. Argi, J. Mol. Catal. A: Chem., 2013, 368–369, 16–23 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXS-97, Rel. 97-2, A Program for Automatic Solution of Crystal Structures, Gottingen University, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL Version 2013/4, A Program for Crystal Structure Refinement, Gottingen University, 2013 Search PubMed.
Footnote |
| † CCDC 957266, 957264 and 957267. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra45546g |
| This journal is © The Royal Society of Chemistry 2014 |