
CO2/oxalate cathodes as safe and efficient alternatives in high energy density metal–air type rechargeable batteries
Abstract
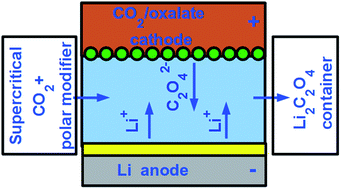
We present theoretical analysis on why and how rechargeable metal–air type batteries can be made significantly safer and more practical by utilizing CO2/oxalate conversions instead of O2/peroxide or O2/hydroxide ones, in the positive electrode. Metal–air batteries, such as the Li–air one, may have very large energy densities, comparable to that of gasoline, theoretically allowing for long range all-electric vehicles. There are, however, still significant challenges, especially related to the safety of their underlying chemistries, the robustness of their recharging and the need of supplying high purity O2 from air to the battery. We point out that the CO2/oxalate reversible electrochemical conversion is a viable alternative of the O2-based ones, allowing for similarly high energy density and almost identical voltage, while being much safer through the elimination of aggressive oxidant peroxides and the use of thermally stable, non-oxidative and environmentally benign oxalates instead.
 Please wait while we load your content...
Please wait while we load your content...

