
Structural architecture and solubility of native and modified gliadin and glutenin proteins: non-crystalline molecular and atomic organization
Abstract
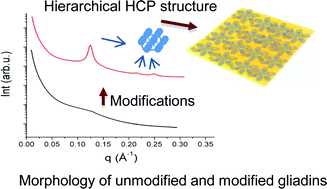
Wheat gluten (WG) and its components, gliadin and glutenin proteins, form the largest polymers in nature, which complicates the structural architecture of these proteins. Wheat gluten, gliadin and glutenin proteins in unmodified form showed few secondary structural features. Structural modification of these proteins using heat, pressure and the chemical chaperone glycerol resulted in a shift to organized structure. In modified gliadin, nano-structural molecular arrangements in the form of hexagonal closed packed (HCP) assemblies with lattice parameter of (58 Å) were obvious together with development of intermolecular disulphide bonds. Modification of glutenin resulted in highly polymerized structure with proteins linked not only by disulphide bonds, but also with other covalent and irreversible bonds, as well as the highest proportion of β-sheets. From a combination of experimental evidence and protein algorithms, we have proposed tertiary structure models of unmodified and modified gliadin and glutenin proteins. An increased understanding of gliadin and glutenin proteins structure and behavior are of utmost importance to understand the applicability of these proteins for various applications including plastic materials, foams, adhesives, films and coatings.
 Please wait while we load your content...
Please wait while we load your content...

