
Sub-2 μm fully porous and partially porous (core–shell) stationary phases for reversed phase liquid chromatography
Abstract
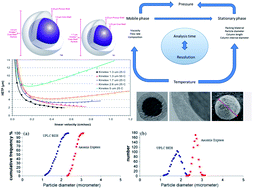
The need for increased throughput and superior performance has increased the demand for stationary phases with improved kinetic performance. Among them, increasing the sample throughput of the ever-growing number of necessary (routine) analyses has become a popular target for reducing precious time. For the past thirty years, high-performance liquid chromatography (HPLC) has been the leading technology when it comes to various analyses; however, the requirement of typically 10–45 min for serial analyses has been a sample throughput-limiting barrier. Recently, the fundamentals of HPLC have been exploited to develop new technologies that can speed up analyses to ground-breaking limits without compromising separation efficiency. This paper reviews the most promising technologies, including porous sub-2 μm ultra-performance liquid chromatography (UPLC) and fused-core particle technology, which have the potential to take LC to the next level. As each analytical method has its own demands, the advances of the above mentioned technologies are discussed for different applications where high throughput analysis can be meaningful. Moreover, we discuss and compare the perspectives of these technologies.
 Please wait while we load your content...
Please wait while we load your content...

