Cobalt nanoparticles in hollow mesoporous spheres as a highly efficient and rapid magnetically separable catalyst for selective epoxidation of styrene with molecular oxygen
Zhi-Qiang
Shi
ab,
Li-Xin
Jiao
ab,
Jian
Sun
ab,
Zi-Bao
Chen
a,
Yuan-Zhe
Chen
ab,
Xiao-Hang
Zhu
ab,
Jing-Hui
Zhou
ab,
Xin-Chun
Zhou
ab,
Xin-Zhe
Li
ab and
Rong
Li
*ab
aState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou, 730000, China. E-mail: liyirong@lzu.edu.cn; Fax: +86 0931 891 2582; Tel: +86 0931 891 2311
bThe Key Laboratory of Nonferrous Metal Chemistry and Resources Utilization of Gansu Province, College of Chemistry and Chemical Engineering Lanzhou University, Lanzhou 730000, China
First published on 6th November 2013
Abstract
Uniform rattle-type hollow mesoporous spheres (HMS) with large cavities have been successfully prepared by the colloidal carbon spheres as templates. The spheres are well monodispersed and nearly uniform in dimension with particle size of ca. 150 nm. Co nanoparticles composed of HMS were found to be a highly efficient catalyst for selective epoxidation of styrene under mild reaction conditions. The catalysts can be easily separated from the reaction mixtures by an external magnet, and reused five times without significant loss of catalytic activity.
1. Introduction
Ordered mesoporous silica materials were first synthesized in 1992.1 Mesoporous spherical self-assembly of nanocarriers have become a promising approach toward the synthesis of ordered mesoporous materials.2–10 As nanocarriers, hollow mesoporous silica materials with unique properties and structures (such as low toxicity, good compatibilities with metal particles, adjustable pore diameter, low density, and very high specific surface area with abundant Si–OH bonds10–12) have attracted much attention as candidates in adsorption and drug delivery systems.2,13–15 The studies showed that the hollow mesoporous silica spheres could provide much higher drug loading capacity, compared to the conventional mesoporous materials such as MCM-41, SBA-15 and MCF. Consequently, the hollow mesoporous spheres have received considerable attention because they have potential applications. For instance, Wang et al. synthesized intact and stable hollow silica spheres with radially oriented amino-functionalized mesochannels, and these hollow spheres showed a much higher release rate and higher flurbiprofen capacity (>1000 mg g−1) compared with flake-like mesoporous SBA-15 particles.16 Zhu et al. demonstrated that hollow mesoporous silica spheres could charge ibuprofen about three times as large as conventional MCM-41 (ref. 17). Yang et al. reported recently gold-promoted palladium catalyst with mesoporous hollow silica spheres as support, and the catalyst showed ten times higher activity than commercial Pd/C catalyst and six times higher activity than Pd/MHSS catalyst.18 Although many catalysts have been found to be reliable, the current chemical transformation emphasizes from environmental and economical concerns require low cost, high efficiency, easy separation and the production of minimum waste. Therefore, more and more researchers are trying to design easily separated and recyclable, environmental and economical catalyst.19–23Epoxide compounds are usually synthesized by epoxidation of alkenes. Therefore, the epoxidation of alkenes is a highly significant chemical reaction. Especially the epoxidation of styrene become increasingly attractive because the epoxide is useful intermediate in the production of many pharmaceuticals and fine chemicals.24 Activation of the carbon–hydrogen bonds of an alkane is considerable difficult due to its stability.25,26 Conventionally, the epoxidation of alkenes was usually carried out with organic peracid as an oxidant or by a chlorohydrin process. A lot of waste was formed in many cases.27,28 Now, organic peroxide and hydrogen peroxide are used as oxidants for the epoxidation of alkenes under the condition of heterogeneous catalyst. For example, Kúzniarska-Biernacka and co-workers found that manganese(III)-Schiff base complex encapsulated in an aluminium pillared clay could efficiently catalyze the epoxidation of styrene by iodosylbenzene (PhIO).29 N.S. Patil and co-workers researched the epoxidation of styrene by anhydrous t-butyl hydroperoxide over Au/Al2O3, Ga2O3, In2O3 and Tl2O3 catalysis.30 Although many current studies about the epoxidation of styrene have been reported,31–34 plenty of difficulties appeared during the reaction, such as low catalytic activity, more byproduct and high reaction temperature. In order to overcome these limitations, researchers exploited environmentally friendly and reusable heterogeneous catalysts for the epoxidation of styrene, such as BaO/Ga2O3,24 Co/Ts-1,34V-MCM-41 (ref. 35) or gold supported on MgO and other alkaline earth oxides.33 Although styrene conversion is quite high when H2O2 or O2 is used as an oxidizing agent, the selectivity for styrene oxide is poor.
Compared with organic peroxides and hydrogen peroxide, molecular oxygen is more environmental and economical oxidant for the epoxidation of styrene. At the same time, developing an efficient catalyst, especially transition metal heterogeneous catalyst for the epoxidation of styrene with O2 is preferable. Herein, we developed a facile route to synthesize HMS nanoreactors with Cobalt nanoparticles resided inside the mesoporous spheres by using the colloidal carbon spheres as the templates. The fabrication procedure involved four steps as shown in Scheme 1. The Co/HMS nanoreactors possess large magnetization, highly open and ordered mesopores, and stably confined. The multicomponent nanostructured materials showed excellent activity for the selective epoxidation of olefins by O2, and could be easily recycled multiple times without visible decrease in catalytic performance. Furthermore, the method is also extended to introduce other transition metal nanoparticles inside the HMS to prepare other new catalysts.
 | ||
| Scheme 1 Scheme of the synthetic procedure for the preparation of Co/hollow mesoporous spheres. | ||
2. Experimental
2.1. Materials and reagents
Glucose, ethanol, tetraethoxysilane (TEOS), ammonia solution (28 wt%), SnCl2·2H2O, N,N′-dimethylformamide (DMF), Co(NO3)2·6H2O. olefins and cetyltrimethy-lammonium bromide (CTAB) Were bought from Alfa Aesar. All Chemicals were of analytical grade and were used as received without further purification.2.2. Preparation of catalysts
2.3. Measurement of catalytic performance
The epoxidation of styrene with O2 was performed in a 25 mL two-necked round bottom flask equipped with a reflux condenser. Typically, 5 mL N,N′-dimethylformamide (DMF) solution containing a given substrate (0.1 mL) and 30 mg catalyst was taken, which ultrasonic treatment for 15 min, Then the mixture was vigorously stirred by a magnetic stirrer and kept in a constant temperature oil bath with the temperature maintained at 100 °C. Thereafter O2 at a certain stable flow rate controlled by a mass flow controller was introduced into the liquid by bubbling at atmospheric pressure. The mixture was vigorously stirred for a specified period of time. Samples were withdrawn by syringe and were analyzed by off-line gas chromatography equipped with a capillary column using flame ionization detector (FID). The catalyst was recovered from the reaction mixture by an external magnetic force, washed with acetone, and dried at 60 °C for 12 hours, and then it was used in the next experiment under the same reaction conditions. We employed the catalyst for other olefins by the same method.2.4. Characterization of catalysts
Powder X-ray diffraction (XRD) patterns were obtained on a Rigaku D/max-2400 diffractometer using Cu-Kα radiation as the X-ray source in the 2θ range of 10–80°. The morphology of the catalyst was observed by a Tecnai G2 F30 transmission electron microscopy and samples were obtained by placing a drop of a colloidal solution onto a copper grid and evaporating the solvent in air at room temperature. The conversion was estimated by GC (P.E. Autosystem XL) and GC-MS (Agilent 6890N/5973N). Co content of the catalyst was measured by inductively coupled plasma (ICP) on IRIS Advantage analyzer. X-ray photoelectron spectroscopy (XPS) was recorded on a PHI-5702 instrument and the C1s line at 297.8 eV was used as the binding energy reference. Magnetic measurement of Co@mesoporous SiO2 nanoreactor was investigated with a Quantum Design vibrating sample magnetometer (VSM) at room temperature in an applied magnetic field sweeping from −15 to 15 kOe.3. Results and discussion
3.1. Characterization of catalysts
Fig. 1 shows the XRD patterns between 10 and 90°. The XRD pattern of Co/HMS exhibits four different peaks at 2θ = 27.1°, 35.2°, 52.4°and 61.5°, corresponding to the reflections of (110), (111), (200) and (220) crystal planes of cobalt, respectively. The pattern shows that the CoO phase is formed by air oxidation during the recovery and storage procedure. | ||
| Fig. 1 XRD patterns of Co/HMS. | ||
The morphologies and structures of the products at different synthetic steps were observed by TEM. Fig. 2(a) shows the TEM micrographs of the carbon nanospheres. It can be clearly observed that the morphology of carbon nanospheres is predominated with spheres; they are quite uniform in size and the average size of the spheres is around 100–150 nm. These carbon nanospheres possess functional groups (–OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) on the surface, which offer an important chemical environment to adsorb metal ion-nanoparticles by electrostatic attraction in acidic solution. Fig. 2(b) is a TEM image of the Co/C composite. The particles with an average size of about 4–6 nm are uniformly distributed on the surfaces of carbon nanospheres. On the basis of Co/HMS using TEOS as the silica source and CTAB as a soft template, a thin layer of mesoporous silica is coated onto the Co/HMS spheres. Then, the Co/HMS nanoreactors were obtained after the calcination aiming at removing the carbon templates and the organic groups of CTAB. Fig. 2(c) shows the TEM micrographs of the calcined HMS in 330 °C. There is a more obvious contrast between the core and the shell of the sphere. A large number of carbon nanospheres' cores began to shrink, but the shell surface is still smooth, no hole formed at the same time. Fig. 2(d) shows the TEM micrographs of the calcined HMS in 430 °C. In contrast to Fig. 2(c), the foam-like masses in the cores have disappeared, which implies that the foam-like masses are organic compounds, and the calcined HMSs has a hollow structure. At the same time, numerous cobalt nanoparticles evenly dispersed on the inner wall of the shell.
O) on the surface, which offer an important chemical environment to adsorb metal ion-nanoparticles by electrostatic attraction in acidic solution. Fig. 2(b) is a TEM image of the Co/C composite. The particles with an average size of about 4–6 nm are uniformly distributed on the surfaces of carbon nanospheres. On the basis of Co/HMS using TEOS as the silica source and CTAB as a soft template, a thin layer of mesoporous silica is coated onto the Co/HMS spheres. Then, the Co/HMS nanoreactors were obtained after the calcination aiming at removing the carbon templates and the organic groups of CTAB. Fig. 2(c) shows the TEM micrographs of the calcined HMS in 330 °C. There is a more obvious contrast between the core and the shell of the sphere. A large number of carbon nanospheres' cores began to shrink, but the shell surface is still smooth, no hole formed at the same time. Fig. 2(d) shows the TEM micrographs of the calcined HMS in 430 °C. In contrast to Fig. 2(c), the foam-like masses in the cores have disappeared, which implies that the foam-like masses are organic compounds, and the calcined HMSs has a hollow structure. At the same time, numerous cobalt nanoparticles evenly dispersed on the inner wall of the shell.
 | ||
| Fig. 2 (a) TEM of carbon nanospheres; (b) TEM of Co/C nanospheres; (c) TEM of final composite nanoreactors (Co/HMS) in low calcined temperature and (d) TEM of final composite nanoreactors in high calcined temperature. | ||
The magnetization curve measured at room temperature for Co/HMS is shown in Fig. 3. The curve presents a thin hysteresis loop, which indicates the ferromagnetic behavior of Co/HMS spheres. The magnetization saturation (Ms) of the mesoporous spheres reaches 5.4 emu g−1, and it suggests that cobalt nanoparticles become encapsulated in hollow mesoporous spheres. This is consistent with the TEM patterns (Fig. 2). Although it is small hysteresis loop, the brown Co/HMS spheres can still be efficiently attracted toward the magnet within a certain period (inset of Fig. 3).
 | ||
| Fig. 3 Room temperature magnetization curves of Co/HMS. | ||
Fig. 4 shows the XPS spectrum of the synthesized Co/HMS nanoreactor. Peaks corresponding to oxygen, carbon, silicon, tin and cobalt are observed. Both the peaks of Si 2s (156.3 eV) and Si 2P (105.5 eV) are quite clear and this means that SiO2 is the main component is the outer surface. However, typical peak of Co elements is not obviously found. To ascertain the oxidation state of the Co, X-ray photoelectron spectroscopy (XPS) studies were carried out. Co 2p photoemission is shown in Fig. 5 for UHV-cleaved CoO (1 0 0) with binding energies of 780.8 eV for 2p3/2 and 795.7 eV for 2p1/2 photoemission maxima, and are in agreement with literature values.38 The majority of cobalt nanoparticles exist inner the hollow mesoporous spheres from the TEM micrographs. Since the analysis of XPS is only about outer surface of particles, the Co 2p signals in Fig. 5 are very small. On the basis of above analysis, it can be concluded that the XPS data further identified the hollow mesoporous structure of the synthesized Co/HMS.
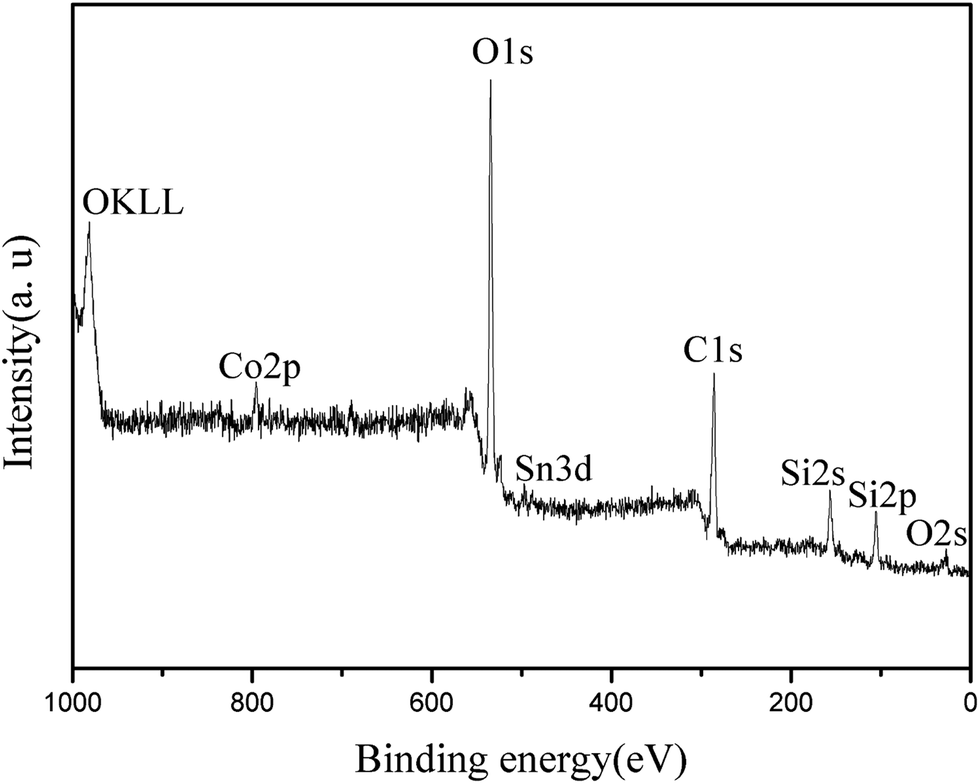 | ||
| Fig. 4 XPS spectrum of the elemental survey scan of Co@mesoporous SiO2. | ||
 | ||
| Fig. 5 XPS spectrum of the Co/HMS showing Co 2p binding energies. | ||
3.2. Epoxidation of styrene reaction catalyzed by Co/HMS nanoreactor
Table 1 compares the effect of solvent on the epoxidation of styrene with O2 over Co/HMS at 100 °C. It can be seen that solvent played an important role on the epoxidation reaction. For example, DMF was especially efficient in obtaining both high styrene conversion and high epoxide selectivity. The conversion date of styrene (Table 1) was obtained in dimethyl sulfoxide (DMSO), toluene and cyclohexanone, but the epoxide selectivity was low.| Solvent | Styrene conversion (%) | Product selectivity (%) | |||
|---|---|---|---|---|---|
| Benab | Epoxide | Pheac | Other | ||
| a Reaction condition: styrene (0.1 mL), catalyst (30 mg), temperature (100 °C), DMF (5 mL), time (3 hours), flow of O2. b Benzaldehyde. c Phenylacetaldehyde. d “—” means none. | |||||
| DMSO | 63 | 8 | 51 | 23 | 18 |
| Chlorobenzene | 0 | —d | — | — | — |
| Cyclohexanone | 49 | 9 | 19 | 24 | 48 |
| Toluene | 34 | 28 | 14 | 6 | 52 |
| DMF | 98 | 0 | 94 | 6 | 0 |
The influence of reaction time on the conversion and selectivity for the epoxidation was showed in Fig. 6. Along with time extension, the conversion of styrene increased rapidly and the epoxide selectivity also increased very markedly (3 hours), and the conversion and selectivity attained over 90%. However as time went by, a growing number of by-products were generated, so the selectivity decreased after 3 hours reaction.
 | ||
| Fig. 6 Influence of reaction time on the epoxidation of styrene by DMF over the Co/HMS (in) catalyst: (a) styrene conversion, (b) epoxidation selectivity. | ||
The compounds were characterized by GC-MS, 1H-NMR spectra of 400 MHz and 13C-HMR spectra of 100 MHz spectrometer and used CDCl3 as the solvent. The chemical shifts (δ) are reported in parts per million (ppm) relative to the residual CHCl3 peak (7.26 ppm for 1H-NMR and 77.0 ppm for 13C-HMR). The coupling constants (J) are reported in Hertz (Hz). Yields refer to isolated material judged to be ≥95% pure by 1H NMR spectroscopy following silica gel chromatography. All the chemicals were used as received unless otherwise stated. 1H-NMR (400 MHz, CDCl3) δ 7.357–7.255 (m, 5H), 3.841 (d, 1H, JXA = 4 Hz, JXB = 2.4 Hz), 3.122 (d, 1H, JAB = 5.6 Hz, JAX = 4 Hz), 2.871(d, 1H, JBA = 5.6 Hz, JBX = 2.4 Hz) shown in Fig. 7. 13C-NMR (100 MHz, CDCl3) δ 128.4, 128.1, 52.2, 51.0 shown in Fig. 8.
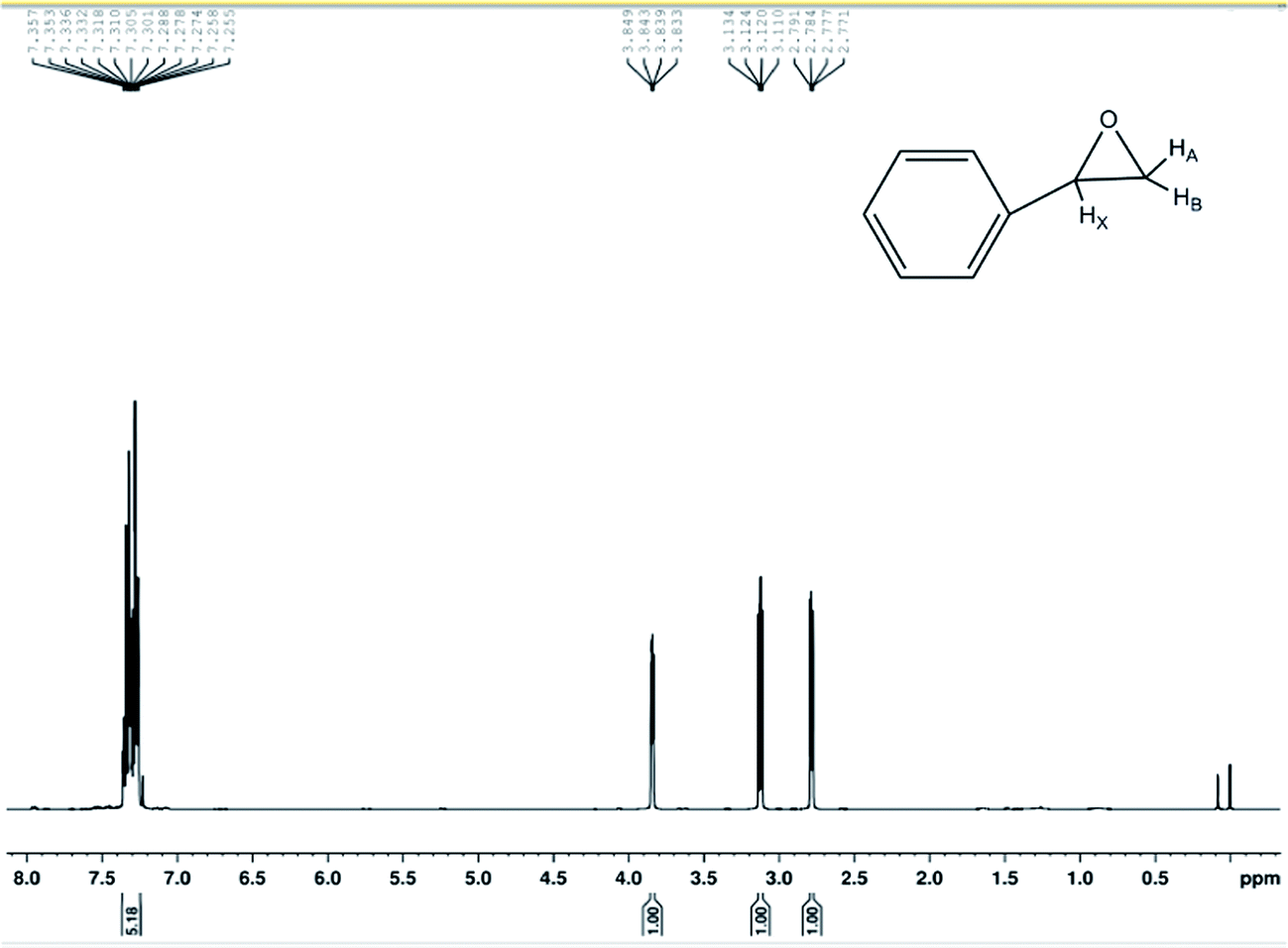 | ||
| Fig. 7 1H-NMR spectra on 400 MHz spectrometer. | ||
 | ||
| Fig. 8 13C-HMR spectra on 100 MHz spectrometer. | ||
We compared the results achieved in this work with the earlier reported heterogeneous catalysis for epoxidation of styrene yield and selectivity (Table 2). Obviously, in our work product yield is the highest. Although the reaction over other catalysts also can receive high yield, they needed more time or higher temperature (Table 2, entry 2–4 and 7–10). Furthermore, some reactions temperature in other works is lower, but the reactions take longer time and obtain lower yield (Table 2, entry 1, 4, 7, 10). By contrast, Co/HMS shows higher conversion and selectivity than entry 3, 5 and 6 in the same time.
| Entry | Catalyst | Solvent | Temp/°C | Time/h | Select/% | Yield/% | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | Co-ZSM-5(L) | TBHP | 90 | 4 | 89 | 87 | 39 |
| 2 | V-MCM-41 | Acetonitrile | rt | 8 | 93 | 77 | 35 |
| 3 | Co–Y–ZrO2 | DMF | 120 | 3 | 90 | 33 | 40 |
| 4 | Cu-MCM-41 | TBHP | 80 | 24 | 80 | 75 | 41 |
| 5 | CuO/Ga2O3 | TBHP | 95 | 3 | 78 | 58 | 42 |
| 6 | Au/Yb2O3 | TBHP | 82 | 3 | 73 | 54 | 43 |
| 7 | Co-SBA-15x | DMF | 100 | 8 | 66 | 62 | 44 |
| 8 | Co3O4 | DMF | 70 | 5 | 84 | 71 | 45 |
| 9 | Co/Ts-1 | DMF | 100 | 3.5 | 74 | 70 | 34 |
| 10 | Co-SSZ-51 | DMF | 100 | 8 | 78 | 37 | 46 |
| 11 | Co/HMS | DMF | 100 | 3 | 94 | 92 | This work |
The catalytic epoxidation of various olefins is summarized in Table 3. Reactions were performed at the optimized reaction temperature under air in different solvent containing alkene, oxidant and catalyst. This catalyst efficiently converts olefins to their corresponding epoxides. Styrene was converted to styrene oxide in high yield. From entry 1, 2 in Table 3, it can be found that carbocyclic alkenes are converted to the corresponding epoxides in high yield, as well. Although oxidation of the linear alkenes requires longer reaction times, the conversion and selectivity are high. All in all, this catalytic system shows a good activity in the case of different olefins.








The catalyst was recycled in order to test its stability as well as activity. The obtained results are showed in Table 4. As seen from the table there was no distinct change observed in conversion, but a slight decrease in yield was observed which presents that the catalysts are stable and five consecutive cycles without any significant loss of catalytic activity.
4. Conclusion
We have developed a facile method for the manufacture of uniform Co/HMS nanoreactor with desired hollow core/mesoporous shell structures, high catalytic activity and magnetic recyclability properties. The cobalt nanoparticles are free of protection ligands and are well-dispersed in mesoporous spheres with little aggregation. The nanoreactors show prominent catalytic activity in epoxidation of styrene with 92% yield for 3 hours and can be recycled five times without significant loss of activity.Acknowledgements
The authors are grateful to Projects in Gansu Province Science and Technology Pillar Program (1204GKCA047), and the Key Laboratory of Catalytic engineering of Gansu Province, China Gansu Province for financial support.References
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- Y. Shang, D. Zhang and L. Guo, J. Mater. Chem., 2012, 22, 856–861 RSC.
- C.-K. Tsung, J. Fan, N. Zheng, Q. Shi, A. J. Forman, J. Wang and G. D. Stucky, Angew. Chem., Int. Ed., 2008, 47, 8682–8686 CrossRef CAS PubMed.
- Y. Fang, D. Gu, Y. Zou, Z. Wu, F. Li, R. Che, Y. Deng, B. Tu and D. Zhao, Angew. Chem., Int. Ed., 2010, 49, 7987–7991 CrossRef CAS PubMed.
- H. Peng, J. Tang, L. Yang, J. Pang, H. S. Ashbaugh, C. J. Brinker, Z. Yang and Y. Lu, J. Am. Chem. Soc., 2006, 128, 5304–5305 CrossRef CAS PubMed.
- Z. Wu, Q. Li, D. Feng, P. A. Webley and D. Zhao, J. Am. Chem. Soc., 2010, 132, 12042–12050 CrossRef CAS PubMed.
- W. Zhou, F. Sun, K. Pan, G. Tian, B. Jiang, Z. Ren, C. Tian and H. Fu, Adv. Funct. Mater., 2011, 21, 1922–1930 CrossRef CAS.
- E. Nilsson, Y. Sakamoto and A. E. C. Palmqvist, Chem. Mater., 2011, 23, 2781–2785 CrossRef CAS.
- J. Chen, Z. Hua, Y. Yan, A. A. Zakhidov, R. H. Baughman and L. Xu, Chem. Commun., 2010, 46, 1872–1874 RSC.
- D. Niu, Z. Ma, Y. Li and J. Shi, J. Am. Chem. Soc., 2010, 132, 15144–15147 CrossRef CAS PubMed.
- Y. Zhu, J. Shi, W. Shen, X. Dong, J. Feng, M. Ruan and Y. Li, Angew. Chem., 2005, 117, 5213–5217 CrossRef.
- L. Y. Xia, M. Q. Zhang, C. e. Yuan and M. Z. Rong, J. Mater. Chem., 2011, 21, 9020–9026 RSC.
- Y. Zhu, T. Ikoma, N. Hanagata and S. Kaskel, Small, 2010, 6, 471–478 CrossRef CAS PubMed.
- X. Yang, S. Liao, J. Zeng and Z. Liang, Appl. Surf. Sci., 2011, 257, 4472–4477 CrossRef CAS.
- L. Guo, J. Li, L. Zhang, J. Li, Y. Li, C. Yu, J. Shi, M. Ruan and J. Feng, J. Mater. Chem., 2008, 18, 2733–2738 RSC.
- J.-G. Wang, F. Li, H.-J. Zhou, P.-C. Sun, D.-T. Ding and T.-H. Chen, Chem. Mater., 2009, 21, 612–620 CrossRef CAS.
- Y. Zhu, J. Shi, W. Shen, H. Chen, X. Dong and M. Ruan, Nanotechnology, 2005, 16, 2633–2638 CrossRef CAS.
- X. Yang, L. Du, S. Liao, Y. Li and H. Song, Catal. Commun., 2012, 17, 29–33 CrossRef CAS.
- G. Yu, B. Sun, Y. Pei, S. Xie, S. Yan, M. Qiao, K. Fan, X. Zhang and B. Zong, J. Am. Chem. Soc., 2010, 132, 935–937 CrossRef CAS PubMed.
- J. Sun, Z. Dong, X. Sun, P. Li, F. Zhang, W. Hu, H. Yang, H. Wang and R. Li, J. Mol. Catal. A: Chem., 2013, 367, 46–51 CrossRef CAS.
- C. Tu, J. Du, L. Yao, C. Yang, M. Ge, C. Xu and M. Gao, J. Mater. Chem., 2009, 19, 1245–1251 RSC.
- Z. Teng, J. Li, F. Yan, R. Zhao and W. Yang, J. Mater. Chem., 2009, 19, 1811–1815 RSC.
- X. H. Xia, J. P. Tu, X. L. Wang, C. D. Gu and X. B. Zhao, Chem. Commun., 2011, 47, 5786–5788 RSC.
- V. R. Choudhary, R. Jha and P. Jana, Green Chem., 2006, 8, 689–690 RSC.
- S. M. Islama, A. S. Roya, P. Mondala, M. Mubaraka, S. Mondala, D. Hossaina, S. Banerjeeb and S. C. Santrab, J. Mol. Catal. A: Chem., 2011, 336, 106–114 CrossRef.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed.
- H.-J. Zhan, Q.-H. Xia, X.-H. Lu, Q. Zhang, H.-X. Yuan, K.-X. Su and X.-T. Ma, Catal. Commun., 2007, 8, 1472–1478 CrossRef CAS.
- J. Sebastian, K. M. Jinka and R. V. Jasra, J. Catal., 2006, 244, 208–218 CrossRef CAS.
- I. Kúzniarska-Biernacka, A. R. Silva, R. Ferreira, A. P. Carvalho, J. Pires, M. B. d. Carvalho, C. Freire and B. d. Castro, New J. Chem., 2004, 28, 853–858 RSC.
- N. S. Patil, R. Jha, B. S. Uphade, S. K. Bhargava and V. R. Choudhary, Appl. Catal., A, 2004, 275, 87–93 CrossRef CAS.
- J. Song, Z. Zhang, T. Jiang, S. Hu, W. Li, Y. Xie and B. Han, J. Mol. Catal. A: Chem., 2008, 279, 235–238 CrossRef CAS.
- Y. Liu, H.-J. Zhang, Y. Lu, Y.-Q. Cai and X.-L. Liu, Green Chem., 2007, 9, 1114–1119 RSC.
- N. S. Patil, R. Jha, B. S. Uphade, S. K. Bhargava and V. R. Choudhary, J. Catal., 2004, 223, 236–239 CrossRef CAS.
- J. Jiang, R. Li, H. Wang, Y. Zheng, H. Chen and J. Ma, Catal. Lett., 2008, 120, 221–228 CrossRef CAS.
- K. M. Parida, S. Singha and P. C. Sahoo, J. Mol. Catal. A: Chem., 2010, 325, 40–47 CrossRef CAS.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 597–601 CrossRef PubMed.
- J. Sun, Z. Dong, P. Li, F. Zhang, S. Wei, Z. Shi and R. Li, Mater. Chem. Phys., 2013, 140, 1–6 CrossRef CAS.
- S. C. Petitto, E. M. Marsh, G. A. Carson and M. A. Langell, J. Mol. Catal. A: Chem., 2008, 281, 49–58 CrossRef CAS.
- B. Qi, X. H. Lu, S. Y. Fang, J. Lei, Y. L. Dong, D. Zhou and Q. H. Xia, J. Mol. Catal. A: Chem., 2011, 334, 44–51 CrossRef CAS.
- B. Tyagi, B. Shaik and H. C. Bajaj, Catal. Commun., 2009, 11, 114–117 CrossRef CAS.
- S. Jana, S. Bhunia, B. Dutta and S. Koner, Appl. Catal., A, 2011, 392, 225–232 CrossRef CAS.
- V. R. Choudhary, R. Jha, N. K. Chaudhari and P. Jana, Catal. Commun., 2007, 8, 1556–1560 CrossRef CAS.
- V. R. Choudhary and D. K. Dumbre, Top. Catal., 2009, 52, 1677–1687 CrossRef CAS.
- H. Cui, Y. Zhang, Z. Qiu, L. Zhao and Y. Zhu, Appl. Catal., B, 2010, 101, 45–53 CrossRef CAS.
- X. H. Lu, Q. H. Xia, D. Zhou, S. Y. Fang, A. L. Chen and Y. L. Dong, Catal. Commun., 2009, 11, 106–109 CrossRef CAS.
- K. M. Jinka, S. M. Pai, B. L. Newalkar, N. V. Choudary and R. V. Jasra, Catal. Commun., 2010, 11, 638–642 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |