Preferential interaction of small-diameter metallic SWNTs with ferroelectric polymer
Mengmeng Ye,
Zhi-Jun Qiu*,
Hui Li,
Minni Qu and
Ran Liu*
State Key Lab of ASIC & System, School of Information Science and Technology, Fudan University, Shanghai, China. E-mail: zjqiu@fudan.edu.cn; rliu@fudan.edu.cn; Fax: +86 21 55664269
First published on 7th April 2014
Abstract
Poly(vinylidene difluoride) (PVDF) blends with single-walled carbon nanotubes (SWNTs) have been prepared to investigate the interaction between nanotubes and polymer. The SWNTs are observed to form a good dispersion in the fluoropolymer matrix. Raman measurements showed sizable changes in the frequencies and intensities of SWNTs and PVDF in the composites compared with the values in pristine SWNTs and neat PVDF. This originates from the polymer's preferential interaction with metallic SWNTs (m-SWNTs), especially with small-diameter tubes, due to non-vanishing Fermi electrons and increased curvature-induced strain energy. In addition, the selective dispersion of m-SWNTs induced remarkable enhancement in the β-ferroelectric phase of PVDF as demonstrated by Fourier transform infrared spectroscopy (FTIR).
Introduction
Poly(vinylidene fluoride) (PVDF) is an important semicrystalline thermoplastic, and has been extensively studied because of its potential applications in piezoelectric, pyroelectric, or electrocaloric materials, which can be used in supercapacitors, actuators, batteries, nonlinear optical fields, and cooling technologies.1–6 It has five different crystalline forms (α, β, γ, δ, ε), in which the nonpolar α- and polar β-phases are the main crystalline polymorphs.7,8 The α-phase has alternating trans- and gauche-bond conformations and is the most common and stable polymorph. The β-phase has an all-trans conformation comprising fluorine atoms and hydrogen atoms on opposite sides of the polymer backbone, which results in a non-zero dipole moment; thus, it exhibits outstanding piezoelectric, pyroelectric, and ferroelectric properties.A variety of experimental techniques have been developed to induce the desirable β-phase formation. The polar β-phase in PVDF is generally obtained by uniaxial or biaxial drawing of α-phase films,9 poling in a strong electric field,10 ultra-fast quenching,11 and crystallization under high pressure.12 Recently, the incorporation of nanofillers has been in focus as a means of enhancing the formation of the β-phase in PVDF.7,13–16 Among the various nanofillers used in PVDF nanocomposites, carbon nanotubes (CNTs) have been considered as ideal reinforcing fillers due to a combination of remarkable electrical, thermal and mechanical properties. Polymer/CNT nanocomposites have been demonstrated with higher stiffness, higher modulus, improved dimensional stability, decreased coefficient of thermal expansion and better electrical conductivity at relatively low CNT concentration.17,18 Due to the zigzag carbon atoms on the CNT surface, which match with the all-trans conformation of β-phase PVDF, the crystallization of PVDF in the β-polymorphic structure is induced. Moreover, the high aspect ratio of CNTs leads to a very low percolation threshold in the polymer nanocomposite.
To our knowledge, although much work on nanocomposites composed of PVDF and multi-walled CNTs (MWNTs) has been reported,19–21 the investigation of single-walled CNTs (SWNTs) as conductive nanofillers is relatively scarce, especially that of the interaction mechanism between SWNTs and PVDF.22 In our work, the dependence of nanotube metallicity on the interface interaction between SWNTs and PVDF has been investigated. It was found that the metallic SWNTs (m-SWNTs) with small diameters are favored to bond with PVDF polymer, resulting in an enhanced ferroelectric β-phase of PVDF.
Results and discussion
As shown in Fig. 1, in contrast to rapid precipitation of SWNTs dispersed in DMF solvent, no settling or segregation of SWNTs in PVDF–DMF solution was observed, even after storing for several months, indicating good dispersion of SWNTs in PVDF–DMF solution. It is worth noting that a homogeneous solution cannot be obtained by adding PVDF solution dropwise to SWNT suspensions. In this case, an excess of SWNTs will exist relative to the polymer, leading to a risk of flocculation. Conversely, if the SWNT suspension is added to the polymer solution, the polymer is in excess during the process. As a result, the polymer chain could adsorb onto the SWNT surface, leading to a stable dispersion. | ||
| Fig. 1 Photographs of SWNTs dispersed in N,N-dimethylformamide (DMF) solvent (a) and SWNTs dispersed in PVDF–DMF solution (b). | ||
The atomic force microscopy (AFM) image of the composite film in Fig. 2 clearly indicates individual SWNTs sparsely embedded in the polymer host. The randomly dispersed bright lines are SWNTs, and bright dots are the ends of the broken SWNTs.23 Yuan et al. suggested that a donor–acceptor interaction occurring between delocalized π-electrons of CNTs and strongly electrophilic F groups of the PVDF chain promotes a homogeneous dispersion of the CNTs in the PVDF matrix through non-covalent bonding.24
 | ||
| Fig. 2 AFM image of SWNTs dispersed in the PVDF host. | ||
Raman spectroscopy with a 514 nm excitation wavelength is used to examine the interaction between the SWNTs and polymer. Fig. 3a shows the low-frequency radial breathing modes (RBM) of the pristine SWNTs and the SWNTs in the composite. The collected spectra are separated and offset vertically for clarity after normalization to the most intense peak. In Fig. 3a, we clearly observed two groups of semiconducting SWNTs (s-SWNTs) near 200 cm−1 and m-SWNTs near 260 cm−1 in the RBM spectra. The characteristics of s- and m-SWNTs were determined from the Kataura plot that is expressed by the relationship between transition energies and nanotube diameter.25 When the energy of the incident laser matches the allowed electronic transitions between the Van Hove singularities of a particular nanotube, Raman signals of the nanotube are resonantly enhanced.
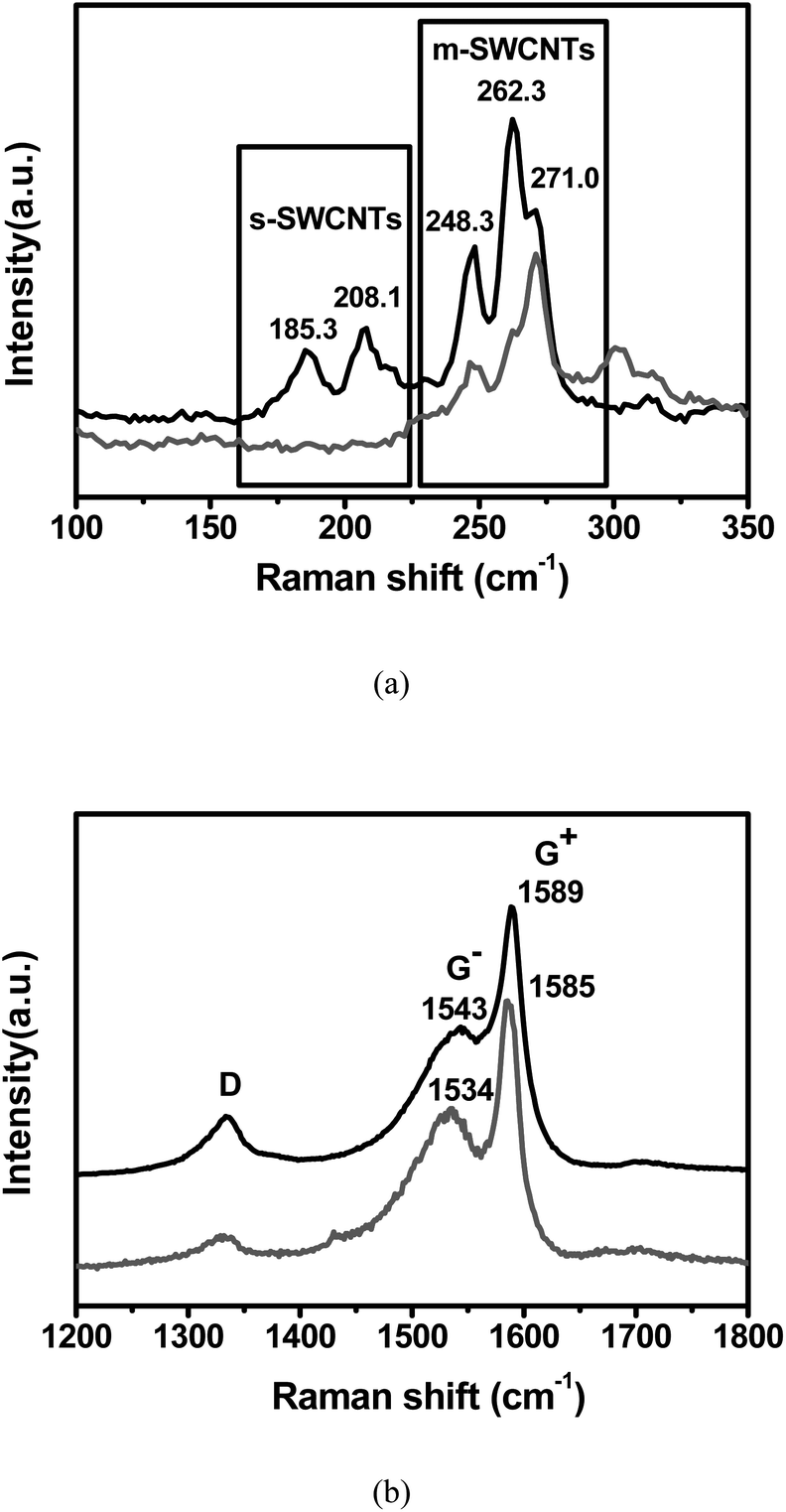 | ||
| Fig. 3 Raman spectra of RBM (a) and G band (b) in pristine SWNTs (black) and PVDF composite (red). | ||
At 514 nm, the excitation is resonant with the ES33 transition in s-SWNTs and the EM11 transition in m-SWNTs. It is found that the two prominent RBM peaks at 185.3 and 208.1 cm−1 from s-SWNTs disappear in the composite, while those peaks at 248.3, 262.3 and 271 cm−1 from m-SWNTs still exist. Interestingly, the relative intensity of the peak at 271 cm−1 becomes large. The diameters (d) of SWNTs were determined by the following relation with Raman shifts (ωRBM):26
 | (1) |
The fraction of m-SWNTs with various diameters is calculated using the ratio of the peak areas, which is obtained through a Lorentzian fit of the RBM peaks. As shown in Table 1, the fraction of small-diameter (0.86 nm) m-SWNTs in the composite is increased two-fold compared to that in pristine SWNTs. The lack of RBM signals from s-SWNTs in the composite was probably caused by the selective removal of s-SWNTs bundles in the centrifugation process, due to stronger van der Waals interaction among the s-SWNTs as compared to s-SWNT–polymer interaction. In contrast, more available charge density at the Fermi level promotes charge transfer from m-SWNTs to PVDF polymer.
| RBM (cm−1)/d (nm) | 271/0.86 | 262.3/0.90 | 248.3/0.95 |
| Pristine SWNTs | 29.3% | 46.8% | 23.9% |
| PVDF/SWNT composite | 58.4% | 24.3% | 17.3% |
Furthermore, the distinct selectivity preferring the small-diameter m-SWNTs can be understood under the mechanism of curvature-induced strain energy in the interaction between PVDF and m-SWNTs. The curvature-induced strain energy in a nanotube per carbon atom, including contributions of pyramidalization angle and π-orbital misalignment, has been shown to be inversely related to the nanotube diameter.27 The higher strain energy in smaller diameter tubes renders them more reactive than larger diameter tubes, which is responsible for the diameter dependence of the interface reaction between nanotubes and PVDF. Similar changes in the RBM frequencies have been observed when SWNTs are functionalized with other polymers.28,29
The strong metallicity-dependent dispersion in the PVDF/SWNT composite was further demonstrated in the Raman spectra of the tangential mode (G band). As shown in Fig. 3b, an intensified and 9 cm−1 downshift of the Breit–Wigner–Fano (BWF) line shape (G− peak) around 1540 cm−1 and the 4 cm−1 downshift of the G+ peak around 1590 cm−1 are observed in the composite compared to the pristine SWNTs. For SWNTs, the G band typically is composed of two separate peaks, G+ and G−. The strong phonon–plasmon coupling in the m-SWNTs results in a downshifted and asymmetric BWF line shape for the G− peak relative to s-SWNTs.30 Moreover, the TO-dominant G+ of metallic tubes is lower than the LO-dominant G+ of s-SWNTs due to the zone folding effect.31 It is also noticed that, accompanied by the shift of the G bands, the intensity of the disorder-induced mode (D band) at 1350 cm−1 in the composite is also greatly reduced as compared with the pristine sample, leading to an increase of the G/D ratio. This behavior seems counterintuitive to other polymer-functionalized SWNTs, where D-band intensity should be enhanced because chemical functionalization disrupted the structure of the SWNTs and introduced more defects in the nanotubes. Our results may be attributed to centrifugal sedimentation, which removes undispersed amorphous carbon, graphitic particles, and other carbon nanoparticles in the composite.
The molecular-level interaction between SWNTs and the PVDF matrix can be further confirmed by changes of the Raman spectra of PVDF, as shown in Fig. 4. The C–H stretch modes around 3000 cm−1 are the only modes of the polymer observed in the Raman spectra. In order to ensure that the treatment in the composite preparation did not produce the changes observed, the polymer without SWNTs was subjected to the same sonication, centrifugation and heat treatment as the composite. It was found that the C–H stretch modes shifted from 2984 and 3024 cm−1 in pristine PVDF to 2979 and 3017 cm−1 in the composite; moreover, the peak at 2979 cm−1 intensified relative to the higher frequency peak. These effects again indicate bonding of the polymer to the SWNTs, especially m-SWNTs with small diameter.
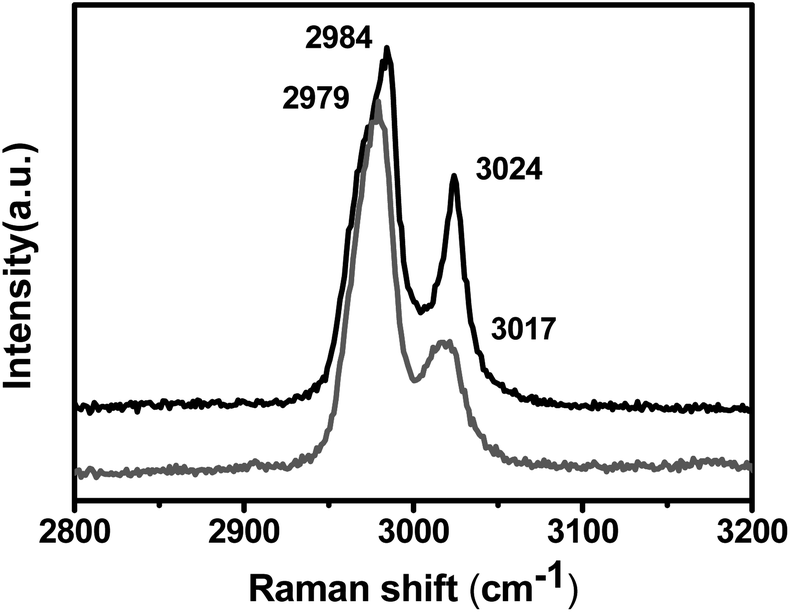 | ||
| Fig. 4 Raman spectra of C–H vibrations in neat PVDF (black) and its composite (red). | ||
Though the crystal lattice energy of the β-phase is slightly lower than that of the α-phase, direct β-phase formation from the solution is prohibited in the PVDF due to the high energy barriers required for transforming the alternating trans- and gauche-bond conformation into the all-trans conformation.32 It was reported that ultrasonic cavitation occurring in the sonication process can generate a local temperature as high as 5000 K, a local pressure as high as 50.6 MPa, and a heating/cooling rate greater than 109 K s−1.33 Under such conditions, PVDF chains are subjected to extremely large forces near collapsing cavitation bubbles. In this process, α-phase polymer chains will obtain energy from the sonicating solution, which can easily overcome the energy barrier for conversion to β-phase. The transformed β-phase molecular chains prefer to be absorbed on the surface of m-SWNTs and act as nucleating agents for the crystallization of polymer chains. The formation of β-phase chains was confirmed by the absorption peaks in the Fourier transform infrared (FTIR) spectra in Fig. 5. In the previous reports, the vibration band at 763 cm−1 is assigned to CF2 bending and skeletal bending of the α-phase, whereas the 840 cm−1 band is ascribed to a mixed mode of CH2 rocking and CF2 asymmetric stretching vibrations in β-phase PVDF. The fraction of β-phase, F(β), can be calculated using the following equation:34
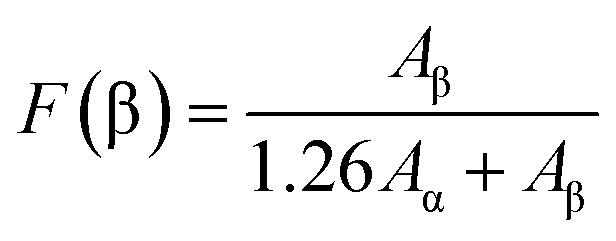 | (2) |
 | ||
| Fig. 5 FTIR spectra of neat PVDF (black) and its composite (red). | ||
Moreover, local piezoelectric hysteresis loops are measured by piezoresponse force microscopy (PFM), in order to demonstrate the ferroelectricity increase in accordance with the β-phase enhancement, as shown in Fig. 6. With the tip bias voltage sweeping, the composite curve shows an obvious hysteresis loop, because of the vertical surface displacement movement, indicating the out-of-plane polarization in the composite film.35 On the other hand, the loop of the pristine PVDF shows small hysteresis, and the local phase responds to the applied tip bias immediately, indicating that the neat PVDF film has almost no ferroelectric nature.
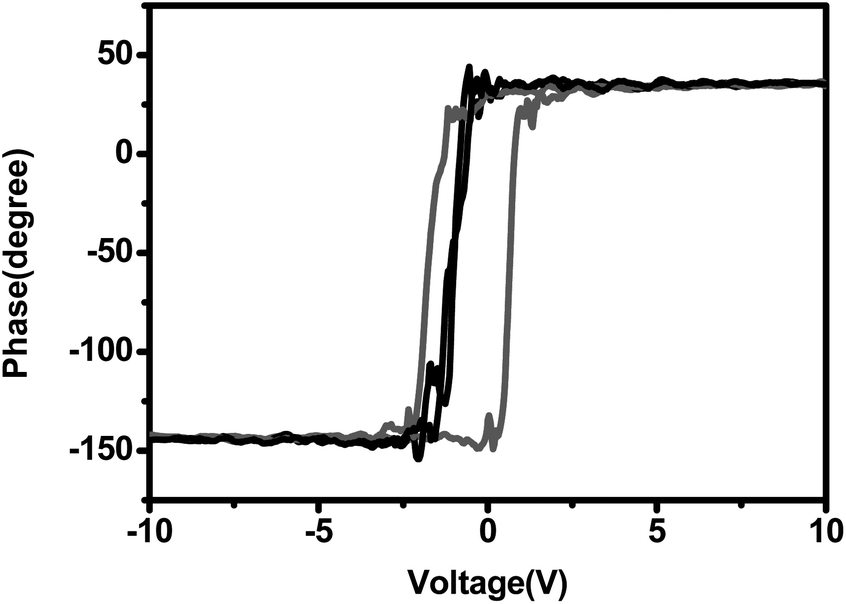 | ||
| Fig. 6 Ferroelectric hysteresis loops of neat PVDF (black) and its composite (red). | ||
Conclusions
PVDF/SWNT nanocomposites have been prepared by a solution sonication method. The SWNTs were observed to form a well-dispersed random nanophase within the fluoropolymer matrix. The Raman spectra show that s-SWNTs are removed and only m-SWNTs are left in the composite. Furthermore, small-diameter m-SWNTs are favored to interact with the polymer due to Fermi electrons and enhanced curvature-induced strain. The selective dispersion of m-SWNTs helps the conversion of PVDF molecules from α- to β-phase during the crystallization of PVDF. The findings in our work are believed to be very significant for the fabrication and applications of PVDF nanocomposites.Experimental section
Materials
PVDF powder (average molecular weight, Mw = 534![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Sigma-Aldrich), HiPco SWNTs (1.1 nm in average diameter and 1.5 μm in average length, Unidym), and N,N-dimethylformamide solvent (DMF, 99.8%, Sigma-Aldrich) were used as received.
000, Sigma-Aldrich), HiPco SWNTs (1.1 nm in average diameter and 1.5 μm in average length, Unidym), and N,N-dimethylformamide solvent (DMF, 99.8%, Sigma-Aldrich) were used as received.
Preparation of PVDF/SWNT composite films
PVDF powder (1 g) was dissolved in 25 ml of DMF solvent at 70 °C for 1 h to ensure complete dissolution, while SWNTs (1 mg) were dispersed in 25 ml of DMF solvent with tip-ultrasonic treatment at the power of 120 W for 1 h. Then, the SWNT suspensions were added dropwise to the PVDF solution with bath-ultrasonic treatment. The resultant mixture was centrifuged at 13000 rpm for 30 min to remove the SWNTs bundles, and the supernatant was drop-cast on a glass dish and kept in oven at 110 °C for 15 min to ensure the removal of the solvent.
Characterization
Atomic force microscopy (AFM) images were obtained via a NanoScope IVA multimode atomic force microscope (DI Dimensions 3100) in tapping mode. The PVDF/SWNT composite films were prepared by drop-casting on a heavily doped silicon substrate.Raman spectra were recorded with a Renishaw inVia confocal micro Raman spectrometer using a 514.5 nm wavelength laser source. To avoid heating the sample, the laser power density was limited to 1 mW μm−2.
Fourier transform infrared (FTIR) spectra were obtained using a Nicolet 6700 spectrometer in the range of 400–4000 cm−1 to obtain the crystal structures for PVDF and nanocomposites.
Piezoresponse force microscopy (PFM) loops were obtained via a NanoScope IVA multimode atomic force microscope (DI Dimensions 3100) to demonstrate the ferroelectricity increase in PVDF/SWNT composite films.
Acknowledgements
This work was financially supported by the Special Funds for Major State Basic Research Projects of China (Grant no. 2011CBA00603), National Natural Science Foundation of China (Grant nos 61171010 and 61204090), Shanghai Municipal Natural Science Foundation (Grant no. 12ZR1402700), and Fundamental Research Project of young teachers to enhance research capacity of Fudan University (no. 20520133248).Notes and references
- S. Manna, A. Mandal and A. K. Nandi, J. Phys. Chem. B, 2010, 114, 2342 CrossRef CAS PubMed.
- Z.-M. Dang, S. H. Yao and H. P. Xu, Appl. Phys. Lett., 2007, 90, 12907 CrossRef PubMed.
- Y. Berdichevsky and Y.-H. Lo, Adv. Mater., 2006, 18, 122 CrossRef CAS.
- E. Peled, T. Duvdevani and A. Melman, Electrochem. Solid-State Lett., 2000, 3, 525 CrossRef CAS PubMed.
- X. Chen, X. Li, X. Qian, M. Lin, S. Wu, Q. Shen and Q. M. Zhang, Polymer, 2013, 54, 5299 CrossRef CAS PubMed.
- X. Li, S. Lu, X. Chen, H. Gu, X. Qian and Q. M. Zhang, J. Mater. Chem. C, 2013, 1, 23 RSC.
- N. Levi, R. Czerw, S. Xing, D. Iyer and D. L. Carroll, Nano Lett., 2004, 4, 1267 CrossRef CAS.
- S. Manna and A. K. Nandi, J. Phys. Chem. C, 2006, 110, 14670 Search PubMed.
- C. A. Nguyen, S. G. Mhaisalkar, J. Ma and P. S. Lee, Org. Electron., 2008, 9, 1087 CAS.
- J. P. Luongo, J. Polym. Sci., 1972, 10, 1119 CAS.
- D. Yang and Y. J. Chen, J. Mater. Sci. Lett., 1987, 6, 599 CAS.
- J. Scheinbeim, C. Nakafuku, B. A. Newman and K. D. Pae, J. Appl. Phys., 1979, 50, 4399 CAS.
- Y. Konishi and M. Cakmak, Polymer, 2006, 47, 5371 CAS.
- H. P. Xu and Z. M. Dang, Chem. Phys. Lett., 2007, 438, 196 CAS.
- G. X. Chen, Y. Li and H. Shimizu, Carbon, 2007, 45, 2334 CAS.
- P. Costa, J. Silva, V. Sencadas, C. M. Costa, F. W. J. van Hattum, J. G. Rocha and S. Lanceros-Mendez, Carbon, 2009, 47, 2590 CAS.
- Z.-M. Dang, L. Wang, Y. Yin, Q. Zhang and Q.-Q. Lei, Adv. Mater., 2007, 19, 852 CAS.
- D. Chen, M. Wang, W. -D. Zhang and T. Liu, J. Appl. Polym. Sci., 2009, 113, 644 CAS.
- Z. Zhao, W. Zheng, W. Yu and B. Long, Carbon, 2009, 47, 2112 Search PubMed.
- X. Tang, M. Hou, L. Ge, J. Zou, R. Truss, W. Yang, M. Yang, Z. Zhu and R. Bao, J. Appl. Polym. Sci., 2012, 125, 592 Search PubMed.
- L. He, Q. Xu, C. Hua and R. Song, Polym. Compos., 2010, 31, 921 CAS.
- R. Shvartzman-Cohen, E. Nativ-Roth, E. Baskaran, Y. Levi-Kalisman, I. Szleifer and R. Yerushalmi-Rozen, J. Am. Chem. Soc., 2004, 126, 14850 CAS.
- L. He, J. Sun, X. Zheng, Q. Xu and R. Song, J. Appl. Polym. Sci., 2011, 119, 1905 CAS.
- J.-K. Yuan, S.-H. Yao, Z.-M. Dang, A. Sylvestre, M. Genestoux and J. Bai, J. Phys. Chem. C, 2011, 115, 5515 CAS.
- H. Kataura, Y. Kumazawa, Y. Maniwa, I. Uemezu, S. Suzuki, Y. Ohtsuka and Y. Achiba, Synth. Met., 1999, 103, 2555 CAS.
- S. M. Bachilo, M. S. Strano, C. Kittrell, R. H. Hauge, R. E. Smalley and R. B. Weisman, Science, 2002, 298, 2361 CAS.
- Z. Chen, W. Thiel and A. Hirsch, ChemPhysChem, 2003, 4, 93 CAS.
- A. Nish, J.-Y. Hwang, J. Doig and R. J. Nicholas, Nat. Nanotechnol., 2007, 2, 640 CAS.
- N. A. Rice, K. Soper, N. Zhou, E. Merschrod and Y. Zhao, Chem. Commun., 2006, 4937 CAS.
- S. D. M. Brown, A. Jorio, P. Corio, M. S. Dresselhaus, G. Dresselhaus, R. Saito and K. Kneipp, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 155414 Search PubMed.
- R. Saito, T. Takeya, T. Kimura, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 57, 4145 Search PubMed.
- B. Mohammadi, A. A. Yousefi and S. M. Bellah, Polym. Test., 2007, 26, 42 CAS.
- S. J. Doktycz and K. S. Suslick, Science, 1990, 247, 1067 CAS.
- A. Salimi and A. A. Yousefi, Polym. Test., 2003, 22, 699 CAS.
- A. Gruverman and S. V. Kalinin, J. Mater. Sci., 2006, 41, 107 CAS.
| This journal is © The Royal Society of Chemistry 2014 |