
Surface-enhanced Raman scattering substrate based on a Ag coated monolayer array of SiO2 spheres for organic dye detection
Abstract
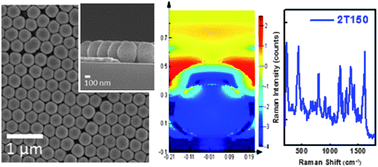
In this study, we developed a facile technique to fabricate a surface-enhanced Raman scattering (SERS) substrate. The technique involves self-assembling monodispersive SiO2 spheres on silicon wafer, then coating with a thin Ag film to form a SERS substrate comprised of a Ag coated monolayer array of SiO2 spheres. The substrate can detect minute amounts of organic dye. By optimizing the fabrication process, including controlling the size of the SiO2 spheres, the distribution of the SiO2 spheres on the silicon wafer, and the thickness of the Ag film, a dramatic increase in the enhancement of Raman scattering signals by surface plasmon resonance can be achieved. The enhancement of Raman scattering is closely correlated to the absorption peak of the organic dye and the excitation wavelength of the Raman system. A 40 000-fold enhancement has been observed for a substrate prepared with 350 nm SiO2 spheres self-assembled on silicon wafer and coated with a 150 nm Ag layer. Through experimental and theoretical calculations (FDTD), such a strong signal intensity originates from the closeness between the absorption peak of the organic dye and the Raman excitation wavelength occurring at the hot spots of the SERS substrate. The high sensitivity, low cost and quick response provided by this type of SERS substrate are useful for the design and fabrication of functional devices and sensors.
 Please wait while we load your content...
Please wait while we load your content...

