Scalable and facile preparation of graphene aerogel for air purification†
Junfei Liangab,
Zhi Caia,
Lidong Lia,
Lin Guo*a and
Jianxin Geng*b
aKey Laboratory of Bio-Inspired Smart Interfacial Science and Technology of Ministry of Education, School of Chemistry and Environment, Beihang University, 37 Xueyuan Road, Haidian District, Beijing 100191, China. E-mail: guolin@buaa.edu.cn; Fax: +86-10-8233-8162; Tel: +86-10-8233-8162
bNational Engineering Research Center of Engineering Plastics, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, 29 Zhongguancun East Road, Haidian District, Beijing 100190, China. E-mail: jianxingeng@mail.ipc.ac.cn; Fax: +86-10-6255-4670; Tel: +86-10-6255-4670
First published on 9th December 2013
Abstract
Amine-functionalized graphene aerogel was prepared through gelation and in situ reduction of GO by using polyethylenimine, and subsequent freeze-drying of the resultant graphene hydrogel. Having a large specific surface area, continuous pore structures, and active chemical adsorbing sites, the graphene aerogel exhibited better properties in adsorbing formaldehyde.
Graphene has received enormous attention due to its intriguing properties and wide potential applications in various fields such as nanoelectronics, transparent conductors, sensors, energy-related materials, and biomedical applications.1 To explore the unique properties and practical applications of graphene-based materials, a prerequisite is mass-production of these materials with controlled composition and microstructures. Developing macroscopic graphene-based materials (MGM) with industrial interest is still a challenge for expanding the practical applications of graphene. To this end, MGM of various forms, i.e. one dimensional (1D), 2D and 3D, have been fabricated and some of the as-prepared MGM demonstrated numerous breakthroughs in corresponding areas.2 Among them, 3D MGM is now receiving unprecedented attention because it not only has the excellent intrinsic properties of graphene but also provides conductive framework with high specific areas for catalyst loading, organic or inorganic molecule absorption, and efficient routes for charge or ion transportation.3 However, it is still a challenge to prepare a large-scaled 3D MGM with engineered properties through a facile method.
Herein, we report a facile one-step solution route for chemical synthesis of 3D macroscopic graphene aerogel (MGA). In this process, polyethylenimine (PEI) was used for (1) its reduction effect,4 (2) potential hydrogen bond interactions with GO, which led to formation of GO hydrogel,5 and (3) adsorption performance of the amine groups for formaldehyde (HCHO).6 The in situ reduction, self-assembly, and functionalization of graphene were realized in one step under a mild condition. In addition, the amine-functionalized MGA turned out to be efficient for adsorption of toxic HCHO. To the best of our knowledge, this is the first report that uses a polyelectrolyte as both reducing reagent and cross-linking reagent for preparing functionalized MGA having air purification capacity. The method presented in this paper provides a facile, scalable, economic, and green strategy for preparation of MGM.
The overall procedure for preparing MGA is illustrated in Scheme 1. First, GO suspension and PEI solution in water were prepared. GO sheets are negatively charged due to the presence of carboxylic acid groups, whereas PEI chains are positively charged due to the presence of the primary, secondary, and tertiary amines which are protonated in a aqueous solution. Second, upon mixing the GO suspension and the PEI solution, the GO sheets and the PEI chains were brought together by electrostatic and hydrogen bond interactions. Around 15 min later, the colour of the suspension changed from brown to black (Fig. S1a and b†), indicating the reduction of GO by PEI. During the reduction process, the graphene sheets decorated with PEI simultaneously self-assembled into a 3D hydrogel through the electrostatic and hydrogen bond interactions.5 In this process, PEI served as a reduction reagent for reducing GO, a stabilizing reagent for preventing aggregation of the resultant graphene sheets, and a cross-linking reagent for forming graphene hydrogel. After a mild reaction at 90 °C for ca. 6 h without stirring, phase separation took place in the reaction vessel (Fig. S1c†): the graphene hydrogel floated on the top of a transparent liquid. Finally, a 3D monolithic architecture of black MGA was obtained by freeze-drying the graphene hydrogel.
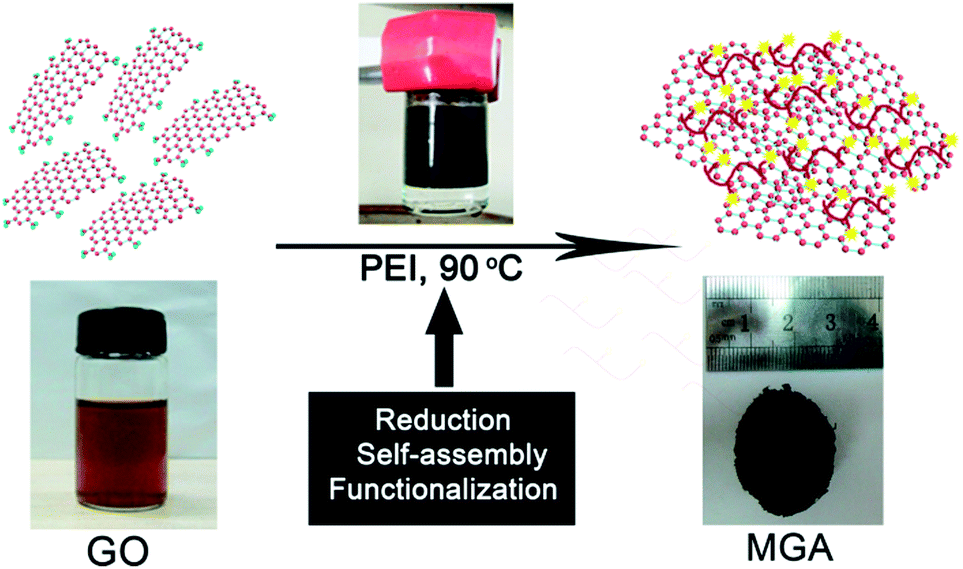 | ||
| Scheme 1 Schematic illustration of fabrication of MGA. | ||
With the MGA in hand, FT-IR spectra, Raman spectra, and X-ray photoelectron spectra (XPS) were collected in order to investigate the reduction of GO by PEI, as well as the interfacial interactions between the resultant graphene sheets and PEI. GO yielded a IR spectrum containing peaks at 1401, 1735, 1220, 1052, and 827 cm−1 corresponding to the vibration of O–H, COOH, C–OH, C–O, and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively, as well as the skeletal vibration of the remaining sp2 domains at 1620 cm−1 (Fig. 1a).7 After mixing of the PEI solution with a GO suspension, GO was reduced, as confirmed by the IR spectrum of MGA, in which the vibration peaks corresponding to oxygen-containing groups of GO disappeared. On the other hand, the skeletal vibration of the sp2 domains was remaining, indicative of the conversion of GO to graphene. In Fig. 1b are displayed the Raman spectra of GO and MGA. The Raman spectrum of GO contained D band and G band at 1359 and 1604 cm−1, whereas the G band in the spectrum of MGA shifted by 16 cm−1 to a lower frequency at 1588 cm−1. Such shift might be due to the charge transfer from the amine groups of PEI to graphene sheets,8 indicating successful hybridization of the graphene sheets by PEI. Furthermore, the intensity ratio of D band to G band for MGA increased compared to that of GO (from 0.93 to 1.19). This is a commonly observed phenomenon for chemically converted graphene because the newly formed myriad sp2 domains decreased the average size of all the sp2 domains upon reduction of the GO sheets.3b In Fig. 1c are displayed the C 1s XPS of GO and MGA. It is seen that the spectrum of GO could be deconvoluted into three components, with two major peaks at 284.7 and 286.8 eV corresponding to the graphitic sp2 C species and C–O species respectively, as well as a minor peak at 287.9 eV due to O–CO and CO species.9 In contrast, the peaks derived from the C species associated with oxygen-containing groups (hydroxyl, epoxide, and carboxylic acid groups) almost disappeared in the XPS of MGA, indicating the efficient reduction of GO by PEI. Additionally, we also found a peak corresponding to C–N species at 285.3 eV in the XPS of MGA, indicating the existence of amine groups in the graphene aerogel, which were significant for the MGA to be used as an adsorbent for removal of HCHO.
O bonds, respectively, as well as the skeletal vibration of the remaining sp2 domains at 1620 cm−1 (Fig. 1a).7 After mixing of the PEI solution with a GO suspension, GO was reduced, as confirmed by the IR spectrum of MGA, in which the vibration peaks corresponding to oxygen-containing groups of GO disappeared. On the other hand, the skeletal vibration of the sp2 domains was remaining, indicative of the conversion of GO to graphene. In Fig. 1b are displayed the Raman spectra of GO and MGA. The Raman spectrum of GO contained D band and G band at 1359 and 1604 cm−1, whereas the G band in the spectrum of MGA shifted by 16 cm−1 to a lower frequency at 1588 cm−1. Such shift might be due to the charge transfer from the amine groups of PEI to graphene sheets,8 indicating successful hybridization of the graphene sheets by PEI. Furthermore, the intensity ratio of D band to G band for MGA increased compared to that of GO (from 0.93 to 1.19). This is a commonly observed phenomenon for chemically converted graphene because the newly formed myriad sp2 domains decreased the average size of all the sp2 domains upon reduction of the GO sheets.3b In Fig. 1c are displayed the C 1s XPS of GO and MGA. It is seen that the spectrum of GO could be deconvoluted into three components, with two major peaks at 284.7 and 286.8 eV corresponding to the graphitic sp2 C species and C–O species respectively, as well as a minor peak at 287.9 eV due to O–CO and CO species.9 In contrast, the peaks derived from the C species associated with oxygen-containing groups (hydroxyl, epoxide, and carboxylic acid groups) almost disappeared in the XPS of MGA, indicating the efficient reduction of GO by PEI. Additionally, we also found a peak corresponding to C–N species at 285.3 eV in the XPS of MGA, indicating the existence of amine groups in the graphene aerogel, which were significant for the MGA to be used as an adsorbent for removal of HCHO.
 | ||
| Fig. 1 (a) FT-IR spectra, (b) Raman spectra, and (c) C 1s XPS of GO and MGA. | ||
Solid-state 13C MAS NMR spectra were used to explore the structural change from GO and MGA. Fig. S2† displays the solid-state 13C MAS NMR spectra of GO and MGA. In the spectrum of GO, the peaks at 61 and 70 ppm represent the 13C nuclei in the epoxide and hydroxyl groups, respectively. The peak at 134 ppm belongs to the un-oxidized sp2 carbons of the graphene network.10 For graphene oxide with a low degree of oxidation, the latter resonances overlap, and two weak broad resonance corresponding to carboxylic carbons and carbonyl carbons are observed at 169 ppm and 190 ppm, respectively.11 In the 13C spectrum of the MGA, the 61 ppm peak, the 169 ppm peak and 190 ppm peak disappeared, and the 70 ppm resonances weaken significantly, which results from the chemical reduction of GO. The peak at 134 ppm shifts to 122 ppm because of the change in the chemical environment of the sp2 carbons.10a,11b As expected, the NMR spectrum of GMA contains the feature resonances of PEI. A very strong signal at 48 ppm can be assigned to the aliphatic CH2 groups of the PEI.12 On the other hand, a peak at 163 ppm corresponding to the carbon in amide is observed,13 which is formed by the reaction between the carboxyl group of GO and the amine group of PEI. Therefore, the NMR data further support that the GO have been reduced to graphene and PEI have been bonded to graphene sheets.
In order to investigate the assembly structures of graphene sheets and PEI in the MGA, X-ray diffraction (XRD) of GO and the MGA were performed (Fig. 2). GO yielded a XRD pattern containing a sharp diffraction peak at 10.2°, corresponding to the layer-to-layer distance between GO sheets (0.87 nm).7 In contrast, for MGA, the sharp diffraction peak disappeared in its XRD pattern, with appearance of a broad peak at ca. 23° attributed to amorphous PEI attached on the surfaces of graphene sheets.14 The inexistence of a sharp Bragg reflection corresponding to the layer-to-layer distance in the XRD pattern indicated the absence of layer-to-layer stacking of graphene sheets in the MGA. This result supported the efficiently assembling of the amine-functionalized graphene sheets in the 3D microstructures of the MGA.
 | ||
| Fig. 2 XRD patterns of GO and MGA. | ||
The morphologies of the MGA were studied using scanning electron microscopy (SEM). As summarized in Fig. 3a and b, the as-prepared MGA exhibited well-defined and interconnected 3D microstructures, with uniformly dispersed micron-sized pores. The walls of the 3D microstructures consisting of stacked graphene sheets were rather thin, indicating effective assembly of graphene sheets. Energy dispersive X-ray spectrometry (EDS) mappings of a piece of graphene sheet showed uniform distribution of N and C (Fig. 3c); this result indicated that PEI was uniformly attached on the surfaces of graphene sheets. Therefore, a conclusion can be drawn based on the results of the SEM observation and EDS mappings that the graphene sheets and PEI were efficiently composited together and the distribution of PEI was uniform on the skeleton of the GMA. Furthermore, the synthetic method presented in this paper also showed advantage in scalable preparation: in Fig. 3d is displayed a centimetre-sized block of MGA that was prepared using scaled-up raw materials in a larger reaction vessel.
 | ||
| Fig. 3 SEM images of MGA (a) at low magnification, and (b) at high magnification; (c) SEM image of a piece of graphene sheet and the corresponding EDS maps for N and C; (d) photograph of a centimetre-sized block of MGA. | ||
N2 adsorption–desorption isotherm of MGA was measured by Brunauer–Emmett–Teller (BET) method (Fig. S3†). Specific surface area of the MGA was obtained to be 139.7 m2 g−1, which is bigger than the value of reported graphene aerogel.14 The greater value of this study is beneficial for our MGA to be used as a adsorbing reagent by providing more adsorption sites. Therefore, the big specific surface area, as well as the porous structure proved by SEM (Fig. 3), facilitates the diffusion of gas molecules in the 3D microstructures of MGA, followed by subsequent adsorption of the gas molecules on the active sites.
Next, we explored the application of our amine-functionalized MGA as adsorbent for absorbing HCHO, which is commonly found in newly decorated buildings, but highly toxic to all animals.15 The amine groups attached on the inner walls of the MGA show preferential interactions with HCHO. In Fig. 4 is displayed a HCHO adsorption curve of the MGA. It is seen that the absorption rate was so fast that it only took ca. 5 min to reach adsorption saturation. At the saturation state, the MGA showed a maximum HCHO adsorption capacity of 2.43 mg g−1, which is higher than that of other aminated carbonaceous materials, e.g. activated carbons and graphite oxide.6 The better adsorbing performance of our MGA should be attributed to the following unique features. First, efficient compounding of GO and PEI, and thereafter the resultant graphene and PEI, provided a large specific area of the MGA, which guaranteed a high quantity of active adsorption sites. Second, introduction of amine groups through PEI improved the HCHO adsorption performance of MGA by providing chemical adsorption sites. Third, the continuous pore structures of the MGA provided fast and versatile transport pathways for the HCHO molecules. Therefore, we can conclude that the better performance of MGA as an adsorbing reagent is due to both rational composition design and elaborate structural fabrication.
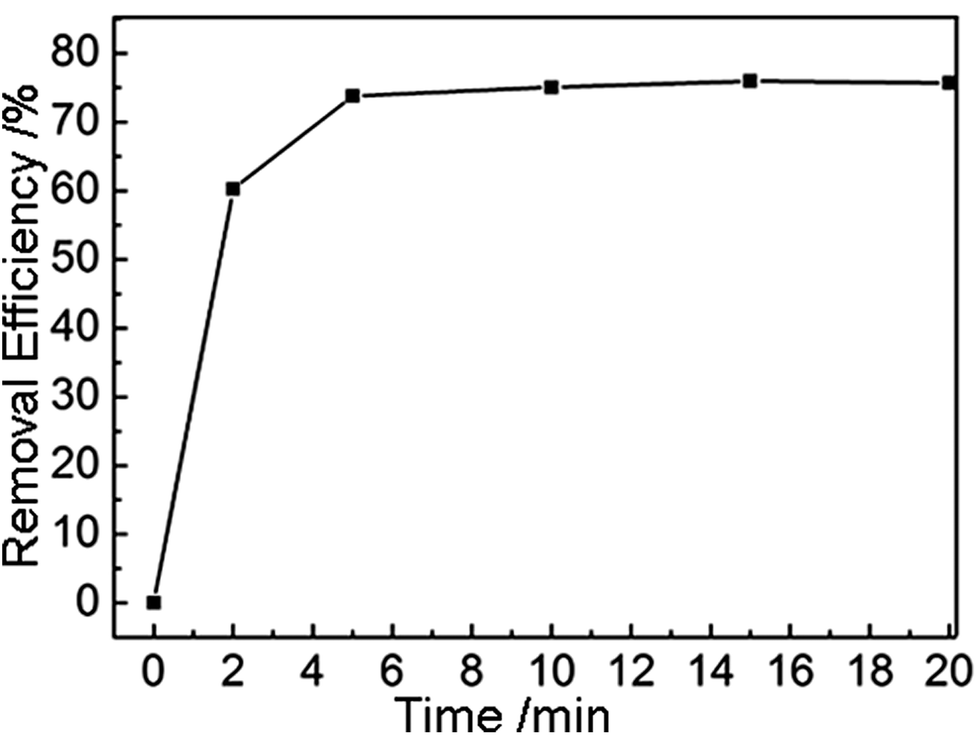 | ||
| Fig. 4 HCHO adsorption curve of the MGA. | ||
Conclusions
In summary, we developed an efficient, economic, and eco-friendly method for preparing MGA through gelation and in situ reduction of GO by PEI, and subsequent freeze-drying of the resultant graphene hydrogel. Such amine-functionalized MGA turned out to be a novel adsorbent for removal of HCHO due to large specific surface area, continuous pore structures, and existence of active chemical adsorbing sites. We expect that our findings will lead to further progress in tailoring the properties of MGA that are potentially applicable in corresponding research areas such as drug/gene delivery, bioimaging, composites, and gas sensors in the future.Materials and methods
Materials
Graphite powder (325 mesh, with purity >99.99%) and branched polyethylenimine (PEI, weight-average molecular weight of 70 kDa) were obtained from Alfa Aesar. All other chemicals (analytical grade) were purchased from Beijing Chemical Co., Ltd. and used without further purification.Preparation of MGA
GO was synthesized from natural graphite powder by the modified Hummer's method.16 The resulting solid graphite oxide was dispersed in water and subjected to dialysis for 7 days to completely remove the residual metal ions and acids. In order to obtain GO nanosheets dispersed in water, the suspension of the graphite oxide was sonicated for 1 h after dialysis. Subsequently, the suspension of GO nanosheets was treated by centrifugation (4000 rpm, 30 min) to precipitate any unexfoliated graphite oxide, and the brown homogeneous supernatant was collected for future use. To prepare MGA, we prepared a mixture of GO and PEI in a 20 mL cylindrical vial with concentrations of 2 mg mL−1 and 1.75 mg mL−1, respectively. The resultant mixture was thermally annealed at 90 °C for 6 h without stirring, leading to phase separation in the vial, with a black graphene hydrogel floating on the top of a transparent liquid. The graphene hydrogel was taken out, washed with distilled water, and freeze-dried, leading to formation of the MGA. Large-sized MGA could be prepared by simply scaling up the preparation.Characterizations
The structures of MGA were characterized by XRD using a Rigaku Dmax 2200 X-ray diffractometer with Cu Kα radiation (λ = 1.5416 Å). The morphologies of MGA were investigated by JEOL JSM-7001F field-emission SEM. Raman spectrometer was recorded on a LabRAM HR800 (HORIBA Jobin Yvon) confocal Raman spectrometer, with an excitation laser wavelength of 488.5 nm. XPS data were collected on an AXIS Ultra instrument Kratos Analytical. IR spectra were recorded using a Nicolet iN10 MX (Thermo Scientific). Specific surface areas were measured at 77 K by BET nitrogen adsorption–desorption process (NOVA 2200e, Quanthachrome, USA.). Solid state 13C CP/MAS NMR spectra were obtained on a Bruker AV-300 spectrometer (spinning speed of 9.5 kHz, recycle delay 5 s, scanning times of 1500–1700) using tetramethylsilane as the internal standard. Chemical shifts are reported in parts per million (ppm).Adsorption of HCHO
The tests for the adsorption of HCHO were performed in a fixed-bed quartz flow reactor using a gas mixture containing 140 ppm HCHO, 20 vol% O2, and balanced He at a total flow rate of 100 cm3 min−1. HCHO standard gas was constantly introduced from a gas-generator system. The effluent concentration of HCHO was monitored using a NEXUS 670-FTIR.Acknowledgements
This project was financially supported by National Basic Research Program of China (2010CB934700), National Natural Science Foundation of China (51272012, and 21274158), the “Hundred Talents Program” of Chinese Academy of Sciences, and Specialized Research Fund for the Doctoral Program of Higher Education (20111102130006).Notes and references
- (a) A. K. Geim, Science, 2009, 324, 1530 CrossRef CAS PubMed; (b) Y. M. Lin, C. Dimitrakopoulos, K. A. Jenkins, D. B. Farmer, H. Y. Chiu, A. Grill and P. Avouris, Science, 2010, 327, 662 CrossRef CAS PubMed; (c) K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706 CrossRef CAS PubMed; (d) G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270 CrossRef CAS PubMed; (e) J. Geng, L. Liu, S. B. Yang, S. C. Youn, D. W. Kim, J. S. Lee, J. K. Choi and H. T. Jung, J. Phys. Chem. C, 2010, 114, 14433 CrossRef CAS; (f) F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652 CrossRef CAS PubMed; (g) Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537 CrossRef CAS PubMed; (h) Z. Liu, J. T. Robinson, X. M. Sun and H. J. Dai, J. Am. Chem. Soc., 2008, 130, 10876 CrossRef CAS PubMed.
- (a) H. P. Cong, X. C. Ren, P. Wang and S. H. Yu, Sci. Rep., 2012, 2, 613 CrossRef PubMed; (b) Z. Dong, C. Jiang, H. Cheng, Y. Zhao, G. Shi, L. Jiang and L. Qu, Adv. Mater., 2012, 24, 1856 CrossRef CAS PubMed; (c) M. F. El-Kady, V. Strong, S. Dubin and R. B. Kaner, Science, 2012, 335, 1326 CrossRef CAS PubMed; (d) Z. Chen, W. Ren, L. Gao, B. Liu, S. Pei and H. M. Cheng, Nat. Mater., 2011, 10, 424 CrossRef CAS PubMed; (e) H. P. Cong, X. C. Ren, P. Wang and S. H. Yu, Energy Environ. Sci., 2013, 6, 1185 RSC; (f) J. E. Kim, T. H. Han, S. H. Lee, J. Y. Kim, C. W. Ahn, J. M. Yun and S. O. Kim, Angew. Chem., Int. Ed., 2011, 50, 3043 CrossRef CAS PubMed.
- (a) C. Li and G. Shi, Nanoscale, 2012, 4, 5549 RSC; (b) H. Bi, X. Xie, K. Yin, Y. Zhou, S. Wan, L. He, F. Xu, F. Banhart, L. Sun and R. S. Ruoff, Adv. Funct. Mater., 2012, 22, 4421 CrossRef CAS; (c) Z. Niu, J. Chen, H. H. Hng, J. Ma and X. Chen, Adv. Mater., 2012, 24, 4144 CrossRef CAS PubMed; (d) Z. S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082 CrossRef CAS PubMed; (e) H. Feng, Y. Li and J. Li, RSC Adv., 2012, 2, 6988 RSC; (f) H. Huang, S. Lü, X. Zhang and Z. Shao, Soft Matter, 2012, 8, 4609 RSC; (g) H. Huang, P. Chen, X. Zhang, Y. Lu and W. Zhan, Small, 2013, 9, 1397 CrossRef CAS PubMed.
- (a) C. Tian, B. Mao, E. Wang, Z. Kang, Y. Song, C. Wang and S. Li, J. Phys. Chem. C, 2007, 111, 3651 CrossRef CAS; (b) X. Sun, S. Dong and E. Wang, Macromolecules, 2004, 37, 7105 CrossRef CAS.
- H. Bai, C. Li, X. Wang and G. Shi, J. Phys. Chem. C, 2011, 115, 5545 CAS.
- (a) Y. Matsuo, Y. Nishino, T. Fukutsuka and Y. Sugie, Carbon, 2007, 45, 1384 CrossRef CAS PubMed; (b) C. Ma, X. Li and T. Zhu, Carbon, 2011, 49, 2873 CrossRef CAS PubMed.
- Y. M. Li, X. J. Lv, J. Lu and J. H. Li, J. Phys. Chem. C, 2010, 114, 21770 CAS.
- M. Zhu, P. Chen and M. Liu, ACS Nano, 2011, 5, 4529 CrossRef CAS PubMed.
- N. L. Yang, J. Zhai, D. Wang, Y. S. Chen and L. Jiang, ACS Nano, 2010, 4, 887 CrossRef CAS PubMed.
- (a) S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS PubMed; (b) X. Wang, Y. Hu, L. Song, H. Yang, W. Xing and H. Lu, J. Mater. Chem., 2011, 21, 4222 RSC.
- (a) W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403 CrossRef CAS PubMed; (b) Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679 CrossRef CAS PubMed.
- E. P. Dillon, C. A. Crouse and A. R. Barron, ACS Nano, 2008, 2, 156 CrossRef CAS PubMed.
- D. Meng, J. Sun, S. Jiang, Y. Zeng, Y. Li, S. Yan, J. Geng and Y. Huang, J. Mater. Chem., 2012, 22, 21583 RSC.
- (a) H. D. Pham, V. H. Pham, T. V. Cuong, T. D. Nguyen-Phan, J. S. Chung, E. W. Shin and S. Kim, Chem. Commun., 2011, 47, 9672 RSC; (b) X. Zhang, Z. Sui, B. Xu, S. Yue, Y. Luo, W. Zhan and B. Liu, J. Mater. Chem., 2011, 21, 6494 RSC.
- (a) E. M. Carter, L. E. Katz, G. E. Speitel and D. Ramirez, Environ. Sci. Technol., 2011, 45, 6498 CrossRef CAS PubMed; (b) G. McGwin, J. Lienert and J. L. Kennedy, Environ. Health Perspect., 2010, 118, 313 CrossRef CAS PubMed.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Photographs of the time-dependent formation process of the MGH and N2 adsorption–desorption isotherms of the MGA. See DOI: 10.1039/c3ra45147j |
| This journal is © The Royal Society of Chemistry 2014 |