Biotransformation of diterpenes
María Rico-Martínez
,
Fernanda G. Medina
,
Joaquín G. Marrero
* and
Soraya Osegueda-Robles
*
Instituto Politécnico Nacional, Unidad Profesional Interdisciplinaria de Ingeniería Campus Guanajuato, Av. Mineral de Valenciana, No. 200, Col. Fracc. Industrial Puerto Interior, C.P. 36275 Silao de la Victoria, Guanajuato, Mexico. E-mail: jgonzalezm@ipn.mx
First published on 4th February 2014
Abstract
Diterpenes are a versatile group of biologically active ingredients present in several phytoextracts. Structural modification of the diterpenes to enhance their pharmaceutical relevance can be efficiently carried out by the application of biotransformational processes using microorganisms or isolated enzymes. Over the past years, special attention has been paid to the biotransformation of diterpenes due to the fact that biocatalysts allow the production of enantiomerically pure compounds using mild and environmentally friendly processes. A wide range of microorganisms have been assessed for these biotransformations and have produced encouraging results, as discussed in this review. This report reviews reactions mediated by fungi, published between 2000 and 2013.
 María Rico-Martínez | María Rico Martínez was born in León, México. She is currently pursuing a Bachelor of Engineering degree in Biotechnology and her undergraduate thesis under the guidance of Prof. Joaquín G. Marrero at UPIIG-IPN. Her current research is focused on biotransformation of natural products. |
 Fernanda G. Medina | María Fernanda González Medina was born in León, México. She is currently pursuing a Bachelor of Engineering degree in Biotechnology and her undergraduate thesis under the guidance of Prof. Joaquín G. Marrero at UPIIG-IPN. Her current research is focused on obtention of biodiesel thought biotechnological process, molecular biology, medical chemistry and synthesis of natural products derivatives. |
 Joaquín G. Marrero | Joaquin G. Marrero obtained a BS in chemistry from the University of La Laguna, Spain, in 1999. He received his Ph. D. degree in organic chemistry from the same university under the guidance of Prof. Lucía San Andrés and Prof. Javier G. Luis. Then he carried out postdoctoral research at University of Reading, UK (with Prof. Laurence M. Harwood) and UNAM, Mexico (with Prof. Alfonso Romo and Prof. Luis D. Miranda) during 2005–2009. In 2010, he began his appointment as Professor at UPIIG-IPN, Mexico. His research interests revolve around medicinal chemistry, with a particular affinity for natural products and analogs. |
 Soraya Osegueda-Robles | Soraya Osegueda obtained a BS in chemistry from the University of Guanajuato, México, in 2003. She received her master's degree in chemistry science in 2004 and Ph. D. degree in chemistry from the same university in 2008. In 2008–2009 she was worked by government in PGJ and educational center UNSIJ, México. In 2010, she began her appointment as Professor at UPIIG-IPN, Mexico. Her research interests revolve around analytical chemistry, with a particular affinity for chromatographic methods. |
1. Introduction and scope
The medicinal use of natural products precedes recorded human history probably by thousands of years. As such, they have proved invaluable in providing compounds, either directly or as leads, for therapeutic purposes, such as antibiotics or chemotherapeutic agents.1Terpenoids are the largest and most widespread class of secondary metabolites; approximately 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds have been identified to date with several new compounds being discovered every year.2 Terpenoids are produced in plant cells via two distinctly localized routes. These pathways are named mevalonate pathway (MVA) and methylerythritol 4-phosphate pathway (MEP), respectively. The MEP pathway provides precursors mainly for the synthesis of mono- and diterpenes, isoprene, carotenoids, the phytohormones gibberellins and abscisic acid, phytol, the side chain of chlorophylls, tocopherols, phylloquinones, and plastoquinones while on the other hand, MVA pathway mainly provides isopentenyl diphosphate, essential for the synthesis of sesquiterpenes, sterols, brassinosteroids, polyprenols, and the moieties used for prenylated proteins.3
000 compounds have been identified to date with several new compounds being discovered every year.2 Terpenoids are produced in plant cells via two distinctly localized routes. These pathways are named mevalonate pathway (MVA) and methylerythritol 4-phosphate pathway (MEP), respectively. The MEP pathway provides precursors mainly for the synthesis of mono- and diterpenes, isoprene, carotenoids, the phytohormones gibberellins and abscisic acid, phytol, the side chain of chlorophylls, tocopherols, phylloquinones, and plastoquinones while on the other hand, MVA pathway mainly provides isopentenyl diphosphate, essential for the synthesis of sesquiterpenes, sterols, brassinosteroids, polyprenols, and the moieties used for prenylated proteins.3
The diverse array of terpenoid structures and functions has ignited interest in their commercial use. Terpenoids exhibit several beneficial effects from a biological perspective, including cancer, and also to have antimicrobial, antifungal, antiparasitic, antiviral, antiallergenic, antispasmodic, antihypercholesterolemic, antidiabetic, antiinflammatory, and immunomodulatory properties.4 Based on these favorable biological activities, terpenoids have therefore received considerable phytochemical and biological attention.5–7
In particular, some diterpenoids with insect growth regulatory activity,8 insect antifeedant,9 or insecticidal activity,10 have been isolated from higher plants. Ferruginol, an abietane diterpene, has shown antifungal and antibacterial activity.11 Communic acids, diterpenes based on a labdane skeleton, has shown promising biological activities: antibacterial, antitumoral, hypolipidemic, and relaxing smooth muscle activities have been reported and reviewed recently.12 They have been also used as building blocks for the semi-synthesis of other interesting bioactive compounds, such as quassinoids, antioxidant abietanes and ambrox, a compound with fixative properties particularly prized by perfumers.13 Also, the clerodane has exhibited a wide range of biological activities. Of particular interest is asmarine A and B14 which showed antiproliferative activity against several human cancer cell lines and, clerocidin,15 a naturally occurring antibiotic which has also shown anticancer and antimicrobial activities. The tricyclic diterpene salvinorin A is a trans-clerodane diterpenoid isolated from the mexican plant Salvia divinorum.16 It acts as a kappa opioid receptor agonist and it is the first non-alkaloid compound acting on this receptor.17 Among this class of compounds of great relevance is taxol.18 This compound has attracted growing attention because of its anticancer activity against several tumor cell lines non responsive to other treatments, such as ovarian and breast cancers, non-small-cell and small-cell lung cancer, and cancers to the head and neck.
According to several authors,19 drugs derived from natural products can function not only as new drugs themselves, but also as lead compounds for chemical modifications that will furnish derivatives with increased better activity and pharmacokinetic properties, new mechanisms of action, and fewer adverse side effects.20 A multidisciplinary approach to drug discovery involving the generation of truly novel molecular diversity from natural product sources provides the best solution to increase the productivity in drug discovery and development. Recently, biocatalysis is gradually becoming an important tool for organic synthesis, especially for the production of derivatives, regio- and enantioselectivity, very difficult tasks to achieve by traditional chemical methods or too expensive to perform.21
The term biotransformation can be applied to a specific modification or interconversion of chemical structures performed by enzymes contained in the cells or by an isolated enzyme. Biotransformation differs from fermentation in which the substrate is converted to a desirable product through a complex cell metabolic pathway.22 There is an increasing body of information about use of biocatalysis for selective conversion of synthetic and natural products to intensify either their biological properties or to lead to new biological activities.23 For instance, the ability of microorganisms to hydroxylate chemically inaccessible sites is a potentially powerful synthetic technique.24 In particular, filamentous fungi contain numerous hydroxylating enzymes with broad specificities, able to which catalyze regio- and stereoselective hydroxylation of nonactivated carbons on a variety of natural and synthetic organic compounds.25 Special attention has been paid to the biotransformation of diterpenes because it allows the production of enantiomerically pure compounds, hemisynthesis intermediates, chiral auxiliaries, and chiral synthons of commercial interest under mild reaction conditions. Therefore, bearing in mind that in the last two decades microbial transformation of terpenes has gained increasing popularity and given the fact that there have been many new developments concerning the biomanipulation of diterpenoids we provide an overview of the most significant advances described in the recent literature. Thus, we have chosen to focus exclusively on diterpene biotransformation reactions from precursor molecules, although we excluded taxanes and gibberellins.
2. Clerodanes
Many of clerodane diterpenoids, especially those of a highly oxygenated nature, display potent insect antifeedant, antifungal, antibacterial, anticancer, and other desirable properties.26 The biological activities and challenging structures of the clerodanes have stimulated much synthetic effort, including microbial transformation, that has culminated in many total syntheses.27In this context, Atta-ur-Rahman and co-workers28 describe the synthesis of oxidated derivatives of clerodane lactone 1 and clerodane methyl ester 2 by the plant pathogen fungus Rhizopus stelonifer. In both processes, the authors obtained the resultant products from a cytochrome P450-catalyzed furan ring oxidation, except in the case of 4 where the oxidation occurred not only in the furan ring but at the less active allylic position of clerodane lactone 1 (Scheme 1).
 | ||
| Scheme 1 Oxidation of clerodane lactone 1 and clerodane methyl ester 2 by Rhizopus stelonifer. | ||
The structure of the reactive intermediate resulting from furan ring oxidation is somewhat ambiguous. Two structures have been proposed: an epoxide or a cis-enedione (Scheme 2). Furan oxidation by P450 enzymes is thought to proceed from one of two general mechanisms. The first one involves the direct formation of an epoxide, while the other involves the addition of the high valent iron(IV)-oxospecies to the π-system of the furan ring to produce a tetrahedral intermediate or cationic σ complex that can rearrange to yield either an epoxide 7 or a zwitterionic intermediate 8. Either intermediate 7 or 8 can rearrange to form a cis-enedione 9.29
 | ||
| Scheme 2 Proposed mechanism for enzymatic furan ring oxidation. | ||
3. Pimaranes
Gibberella fujikuroi was used in the microbiological transformation of ent-pimara-7,15-diene diterpenes.30 In these biotransformations, C-19 oxidation did not occur, and the main reaction was the epoxidation of the 7,8-double bond, followed by rearrangement to afford 7-oxo derivatives (Scheme 3). These results were independent of the hydroxylation of C-2, and the angular methyls C-18 and C-19. This indicates a lack of specificity of the enzymes involved in these processes. | ||
| Scheme 3 Microbiological transformation of 2α,19-dihydroxy-9-epi-ent-pimara-7,15-diene 10a and 18-hydroxy-9-epi-ent-pimara-7,15-diene 10b by G. fujikuroi. | ||
To change the double bond from C7–C8 to C9–C11, the reaction product was the epoxidation of the 9(11)-double bond, followed by rearrangement to afford allylic alcohols,31 while in previous work, in the biotransformation of the ent-pimara-7,15-diene derivatives with this fungus,30 the rearrangement of the 7,8-epoxide led to 7-oxo derivatives (Scheme 4). The results of the biotransformations are also influenced by the oxidative state of the methyl-19. Thus, in the incubation of the 13-epi-ent-pimara-9(11),15-diene-19-oic acid 19 (Scheme 4), a double oxidation at C-1 is produced, forming an oxo group, followed by a hydroxylation at C-2, either α or β, to give the corresponding hydroxy ketones 25 and 26, or by a Baeyer–Villiger oxidation to afford the lactone 27, while in the feeding of 19-hydroxy-13-epi-ent-pimara-9(11),15-diene 20, two 7-oxo derivatives were obtained, 22 and 23. The formation of a 1-oxo-2-hydroxy group in the biotransformation of 19 is very similar to that produced in the incubation of some 7-oxo-ent-kaur-16-ene derivatives to give the corresponding 7-oxo-6β-hydroxy derivatives.32 The formation of the lactone 27, represents the first time that a Baeyer–Villiger oxidation has been observed in a microbiological transformation with the fungus G. fujikuroi. It is also worth pointing out that during this enzymatic oxidation process, the “abnormal” lactone resulting from the migration of the less-substituted carbon atom is formed, while the same reaction using chemical reagents occurs at the most substituted position.
 | ||
| Scheme 4 Microbiological transformation of 13-epi-ent-pimara-9(11),15-diene-19-oic acid 19 and 19-hydroxy-13-epi-ent-pimara-9(11),15-diene 20 by G. fujikuroi. | ||
In the last few years, the development of enzymatic methodologies using Baeyer–Villiger monooxygenases (BVMOs) has allowed the preparation of several compounds that are of high interest in organic synthesis. These flavoproteins are oxidoreductases able to catalyse the Baeyer–Villiger oxidation as well as other oxidative processes which employ atmospheric oxygen as the natural oxidant.33 The regioselectivity of the Baeyer–Villiger oxidation is established by steric, conformational and electronic effects leading to the migration of the higher substituted (the most nucleophilic) carbon centre. Nevertheless, in some rare cases the use of Baeyer–Villiger monooxygenases has led to the formation of unexpected lactones with high regioselectivities, formed by the migration of a less-substituted carbon atom,34 increasing the synthetic potential of this class of enzymes.
Ambrosio and co-workers describe the biotransformation of two ent-pimaradienes metabolites isolated from the dichloromethane root extract of Viguiera arenaria:35 ent-pimara-8(14),15-dien-19-oic acid 29 and ent-8(14),15-pimaradiene 30. The incubation of 29 with Glomerella cingulata36 afforded the bioreduction in the carboxylic acid moiety to an alcohol group as sole biotransformation product 31, while its fermentation by Mucor rouxii yielded the isomerization of the endocyclic double bond 32 and oxidation at C-7 33 as the main reaction products.36 When ent-8(14),15-pimaradiene 30, a substrate without the carboxylic acid moiety at C-19, was transformed in the presence of Aspergillus ochraceus, five hydroxylated pimarane-type diterpenes were obtained.37 The main transformations were the stereoselective hydroxylations at C-3, C-7 and C-11 in low yield. In this case, the orientation of hydroxyl group at C-3 was α in the four products (Scheme 5). Although the number of products in each biotransformation is smaller, these results are in agreement with the previously reported observation by Fraga and co-workers.30–32
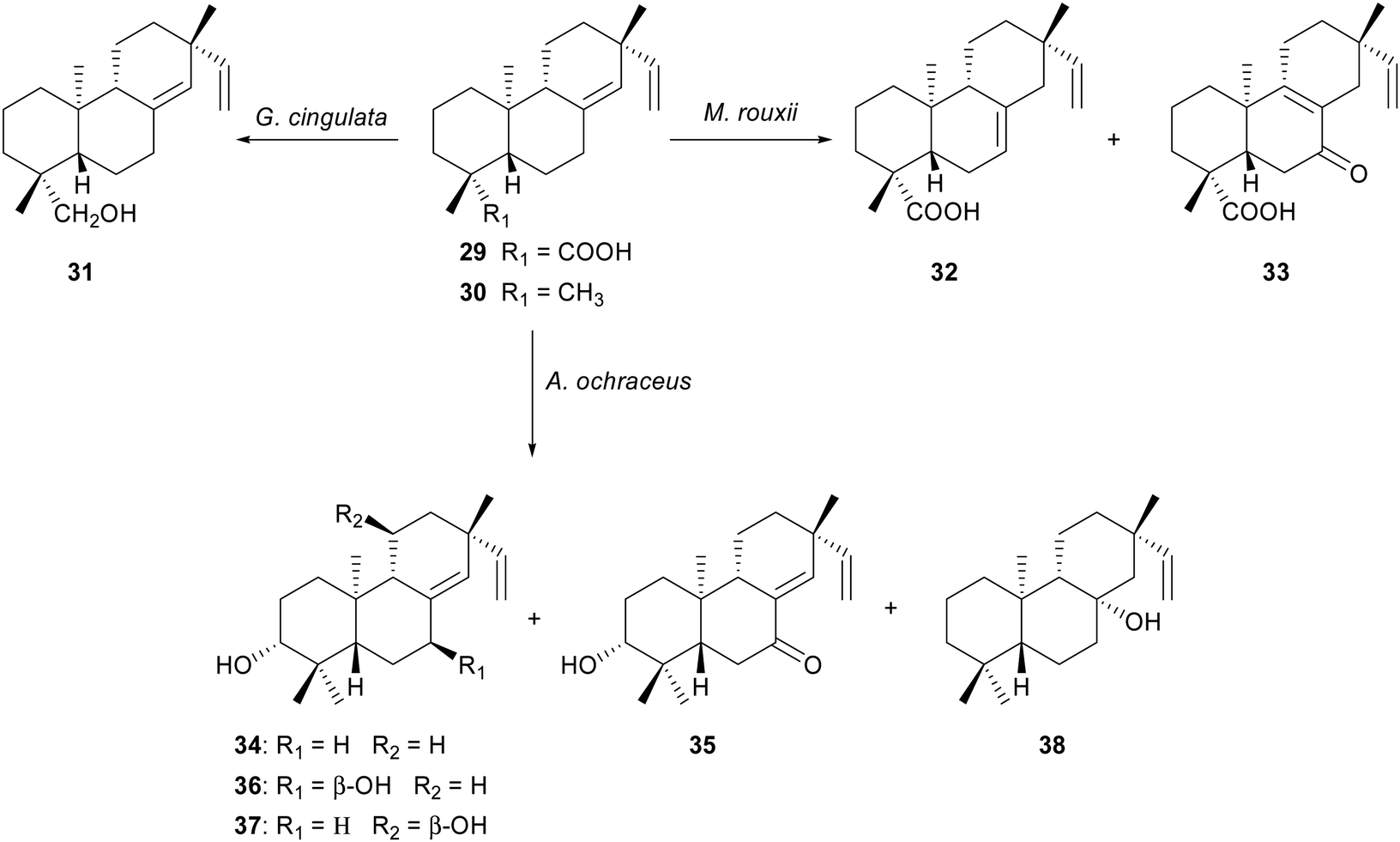 | ||
| Scheme 5 Biotransformation of ent-pimara-8(14),15-dien-19-oic acid 29 and ent-8(14),15-pimaradiene 30. | ||
The incubation of the hydrocarbon 9,13-epi-ent-pimara-7,15-diene diterpene 39, obtained from its 18-hydroxy derivative by treatment with Ph3P/CCl4 and subsequent reduction with tri-n-butyltin hydride, with the fungus G. fujikuroi afforded 1α,9α-dihydroxy-7α,8α-epoxy-13-epi-ent-pimara-15-ene 40 as sole biotransformation product (Scheme 6).38 The main difference between the microbiological transformations of the 18-alcohol (18-hydroxy-9,13-epi-ent-pimara-7,15-diene)30 and the hydrocarbon 39, lies in 39 the rearrangement of the 7α,8α-epoxygroup to give 7-oxo-derivatives was not observed. However, the sequence of reactions 7α,8α-epoxidation, hydroxylation at C-9(α) and subsequent C-1(α) hydroxylation occurred in both incubations.
 | ||
| Scheme 6 Biotransformation of 9,13-epi-ent-pimara-7,15-diene 39 by G. fujikuroi. | ||
4. Abietanes
4.1. Dehydroabietic acid derivatives
Diterpene resin acids are important defense compounds of conifers against potential herbivores and pathogens.39 Dehydroabietic acid 41, one of the major tricyclic diterpenoid constituents of pine resin, exhibits a broad spectrum of biological action. Several activities like antiulcer, antimicrobial, anxiolytic, antiviral, antitumor, anti-inflammatory and cytotoxic have been reported.40 Recent studies have demonstrated that dehydroabietic acid 41 and some derivatives are chemical modulators, particularly openers, of large-conductance calcium-activated Kþ channels (BK channels).41 This feature makes dehydroabietic acid a new scaffold in the treatment of acute stroke, epilepsy, asthma, hypertension, gastric hypermotility and psychoses. Also, dehydroabietic acid 41 was reported to have properties of enhancing the inhibitory activity of anticancer drugs in cervical carcinoma cells, hepatocellular carcinoma cells, or breast cancer cells. This broad spectrum of biological activities, indicate that the compound is a potentially useful starting material for the synthesis of industrial or pharmacologically important products. It is important to note that the microbial degradation or conversion of abietic acid has been scarcely studied due to its chemical lability, causing it to change readily into dehydroabietic acid.42Häkkinen and co-workers assayed the bioconversion of dehydroabietic acid 41 in two plant species: Nicotiana tabacum and Catharanthus roseus cells,43 where dehydroabietic acid 41 can be considered as a xenobiotic for these species, so both of the tested plant species were able to take up and modify this compound according to the typical detoxification pattern of each species. Such is one of the few examples about degradation or bioconversion of dehydroabietic acid 41 using plant cells. Nicotiana tabacum converted dehydroabietic acid 41 into the corresponding 18-O-glycoside 42 (Scheme 7). Madagascar periwinkle (Catharanthus roseus), which endogenously possesses the terpenoid biosynthesis machinery, converted the substrate into two bioconversion products. The first appearance was identified as a 17-hydroxy-dehydroabietic acid 43, following the glycosylation by putative glycosyltransferase into dehydroabietic 17-O-glucoside 44. Hydroxylation of dehydroabietic acid 41 follows a typical detoxification pattern of organic compounds.44 Existing evidence indicates that hydroxylation is the dominant mechanism of resin acid degradation by fungi,45 while several studies suggest that the main enzymes responsible for the oxidative reactions of xenobiotics in plants are cytochrome P450s.46
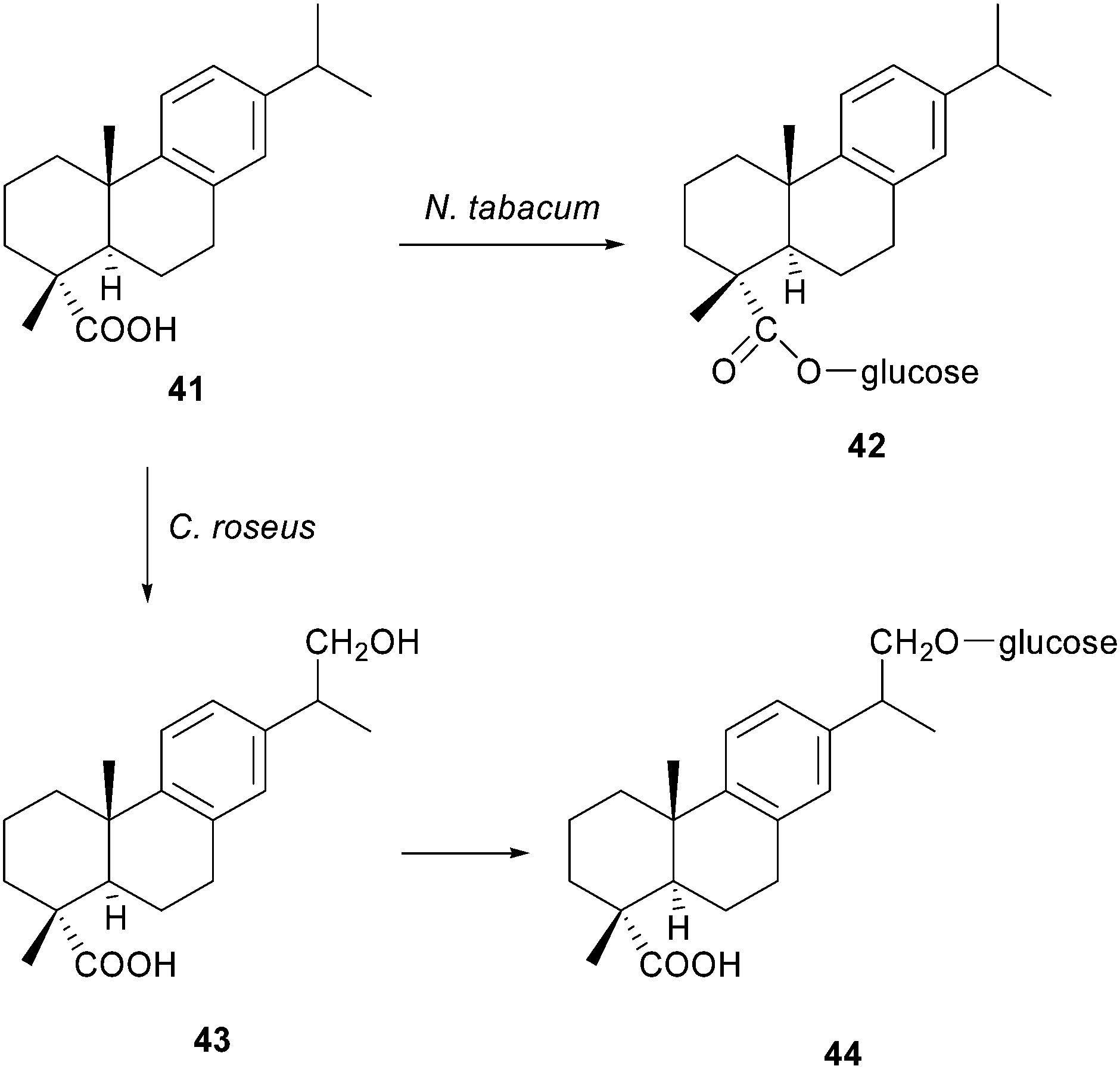 | ||
| Scheme 7 Biotransformation of dehydroabietic acid 41 by N. tabacum and C. roseus cells. | ||
Stereoselective hydroxylation at the C-1 position appeared to be the first degradation step of dehydroabietic acid 41 by two fungi: Trametes versicolor and Phlebiopsis gigantea (Scheme 8).47 This hydroxylation at the C-1 position, has only been previously reported in cultures of two different Fusarium species48 and Aspergillus niger.49 The C-7 and/or C-16 hydroxylations produced in the incubation of dehydroabietic acid 41 with T. versicolor and P. gigantea have also been observed in the substrate biotransformation by the fungi C. cochliodes,45b A. niger,49 F. annosus,50 and M. isabellina.51 Similarly, hydroxylation at C-7 has been reported in studies with the aerobic bacteria Alcaligenes sp., Pseudomonas sp.52 and Pseudomonas abietaniphila.46b,53 Further oxidation of the hydroxyl group at C-7 to a carbonyl function, has equally been observed in studies of the conversion of dehydroabietic acid 41 by the aerobic bacteria Flavobacterium resinovorum54 and Moraxella sp. (HR6).46a A possible pathways for the degradation of dehydroabietic acid 41 is shown in the Scheme 8.47
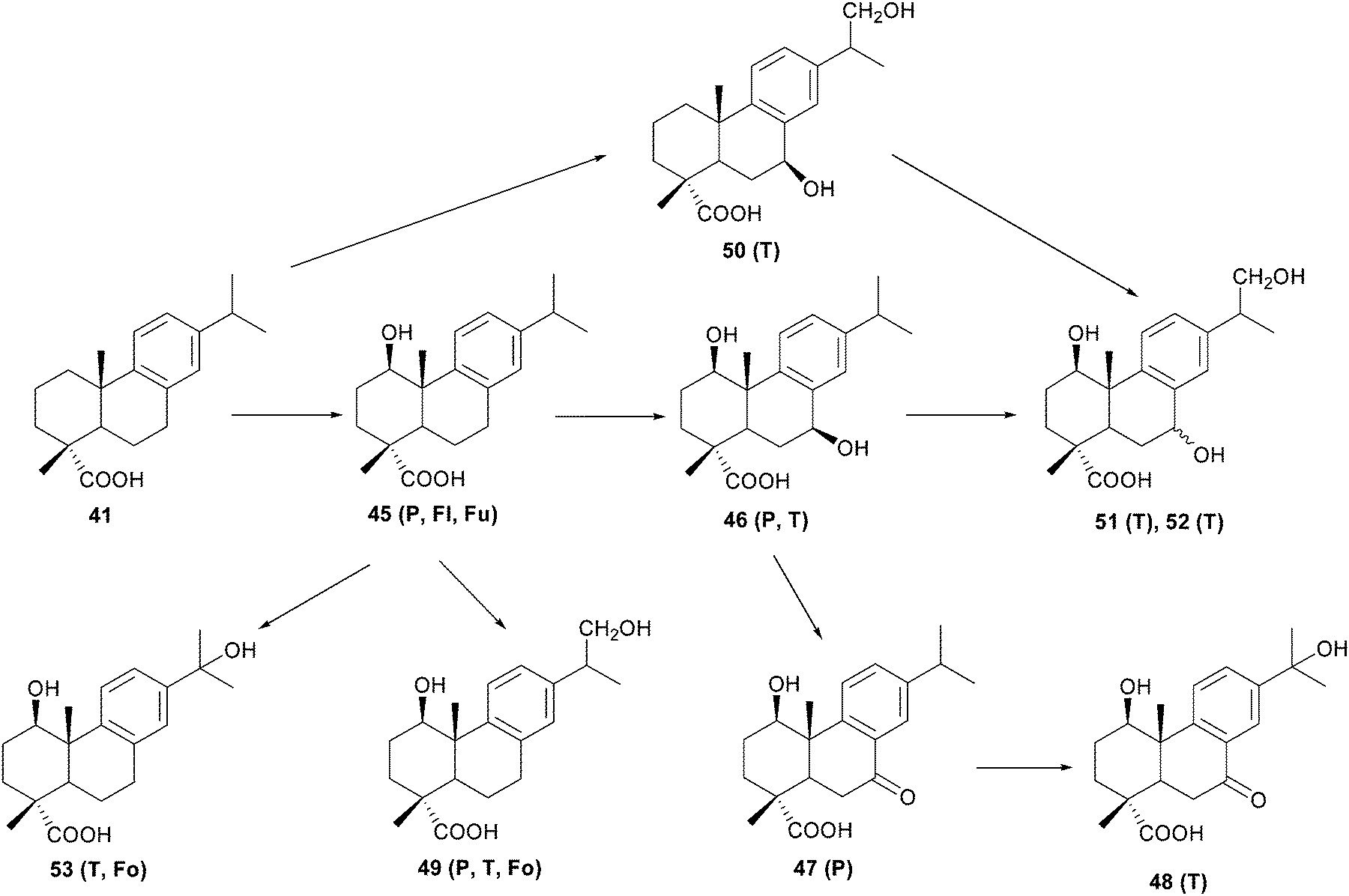 | ||
| Scheme 8 Possible pathways for the degradation of dehydroabietic acid 41 by Trametes versicolor (T), Phlebiopsis gigantea (P), Flavobacterium resinovorum (Fl), Fusarium oxyosporum or F. moniliforme (Fu) and Fomes annosum (Fo).47 | ||
Nagasawa and co-workers have surveyed microorganisms to catalyze the hydroxylation of dehydroabietic acid 41.46a Among 238 microorganisms from a soil sample, three bacteria [(HR1), Moraxella sp. (HR6), and Pseudomonas sp. (HR34)] and two molds [Mucor circinelloides (IT 25) and Mortierella isabellina (HR32)] exhibited dehydroabietic acid 41 derivative-converting activity. The substrate was converted regio- and stereo-selectively by Mucor circinelloides and Mortierella isabellina to give 2α-hydroxydehydroabietic acid 54 (Scheme 9).
 | ||
| Scheme 9 Microbial conversion of dehydroabietic acid 41. | ||
Dehydroabietanol 56 and teideadiol 57 are two abietratriene diterpenes obtained from Salvia pomifera55 and Nepeta teydea56 respectively. The microbiological transformation of both products by Mucor plumbeus57 led to 2α and 7β as the main hydroxylated products (Scheme 10). The core difference between both biotransformations is the 15-hydroxylation of dehydroabietanol 56, which does not occur in teideadiol 57. This could be due to the 1α-alcohol in the latter inhibits the C-15 functionalization. In the incubation of dehydroabietanol 56 with Mucor plumbeus, the 15- and 16-hydroxylations were produced, as in the biotransformation of dehydroabietic acid 41 by Chaetomium cochliodes, Fomes annosus and Mortierella isabellina,47 thus implying a similar functionalization carried out by enzymes from four different genera of fungi.
 | ||
| Scheme 10 Biotransformation of dehydroabietanol 56 and teideadiol 57 by Mucor plumbeus. | ||
4.2. Carnosic acid and carnosol
Carnosic acid 67, an O-diphenolic abietane diterpene precursor of phenolic diterpenes featuring γ- and δ-lactone structures, and related metabolites, such as carnosol 68, are the main compounds responsible for the distinctive antioxidant activity of the popular Labiatae herbs, rosemary and sage.58 Many researchers suggested an oxidation and isomerization pathway for this transformation via an O-quinone intermediate.59 Furthermore, it is suggested that these oxidation pathways were closely related to their antioxidant mechanism. This reactivity was used by San Andrés and coworkers to develop efficient transformations to obtain the minor biologically active abietatriene diterpenes in significative quantities from carnosol 68.60 In addition to their strong antioxidant character, carnosic acid 67 and carnosol 68 exert potent anti-inflammatory and anticarcinogenic properties.61 These compounds inhibit cytochrome P450 activation of carcinogens in human cells in vitro62 and enhance the activities of conjugating enzymes involved in carcinogen detoxification pathways in vivo.63Rosazza and coworkers examined microbial transformations of carnosic acid 67 as a mean of obtaining novel derivatives.64 They screened 49 microorganisms, and only Nocardia sp. (NRRL 5646) was capable of catalyzing the bioconversion of 67, to produce three major metabolites (Scheme 11). In the mechanism proposed by the authors, the Nocardia carboxylic acid reductase reduces the carboxylic acid to an aldehyde group via a carbonyl-activated acyl-adenylate intermediate.65 In whole cell Nocardia cultures, a separate NADPH-dependent alcohol oxidoreductase reduces aldehydes to the corresponding alcohols. The methoxyl group at position C-12 in 71 is likely introduced by means of an S-adenosylmethionine-dependent catechol-O-methyl transferase system.66 The conversion of carnosic acid 67 to carnosol 68 likely involves enzymatic oxidation of 67 to a quinoid intermediate followed by an intramolecular Michael addition of the carboxylate anion at position C7.
 | ||
| Scheme 11 Metabolites formed in the biotransformation of carnosic acid 67 by Nocardia sp. | ||
4.3. Cryptotanshinone
Chemically, tanshinones are 20-norditerpenes with an abietane-type skeleton and a common ortho- or para-naphthoquinone chromophore in the C-ring, which represent the major chemical constituents present in the lipophilic extract of the rhizome of Chinese sage Salvia miltiorrhiza Bunge, a well-known chinese herb used in traditional medicine,67 generally called Danshen.68 Tanshinones share many clinical effects including inhibition of growth in lung cancer tumors,69 atherosclerosis treatment70 aldose reductase inhibitory activity,71 neuroprotective effects,72 apoptosis induction73 and leishmanicidal and antiplasmodial activities.74Cryptotanshinone 73 is one among more than 50 compounds of tanshinones and an active component of S. miltiorrhiza Bunge. Cryptotanshinone 73 was previously shown to possess the most powerful antibacterial activity among the tanshinones, and inhibits the growth of the androgen-independent prostate cancer cell line in vitro and in mice.75 It is an effective inhibitor of topoisomerase I76 and exhibit significant cytotoxicity against a number of cultured human tumor cell lines.77
The biotransformation of cryptotanshinone 73 by Cunninghamella elegans yielded a pair of epimeric alcohols at C-3 and a third product where the unactivated methyl-18 group was oxidized (Scheme 12).78 These biotransformed metabolites are identical to those formed in vivo, in rat bile sample after intravenous administration, which has demonstrated that this fungal biotransformation system could be used to predict and synthesize the mammalian drug metabolites.
 | ||
| Scheme 12 Biotransformation of cryptotanshinone 73 by C. elegans. | ||
The use of microorganisms for simulating the mammalian metabolism of many molecules of pharmacological importance is well documented. Smith and Rosazza postulated the concept of using microorganisms as models for mammalian metabolism of variety of xenobiotics in regio- and stereo-selective manners that are similar to those in mammalian enzyme systems, using both phase I (oxidative) and phase II (conjugative) biotransformation mechanisms.79
The use of microbial models surpass those of animals and offer a number of advantages mainly: (1) simple, easy, can be prepared at low cost. (2) Screening for a large number of strains is a simple repetitive process. (3) The large amount of metabolites formed allows easier detection, isolation, and structural identification. (4) Novel metabolites can be isolated. (5) Useful in cases where regio- and stereo-specificity is required. (6) Maintenance of stock cultures of microorganisms is easier and less expensive than those of cell or tissue cultures or laboratory animals. (7) More reliable and reproducible.80
5. Trachylobane-type diterpenoids
Trachylobane-type diterpenoids are characterized by a pentacyclic carbon skeleton with a tricycle[3.2.1.0]octane system, closely related to the ent-kaurene series. It has been shown that ent-trachyloban compounds possess cytotoxic effects on HeLa and HL-60 cells, and was able to induce apoptosis in human promyelocytic leukemia cells, anti-microbial, anti-tumor, trypanocidal, antifeeding and anti-HIV.81With the aim of obtaining some evidence about the transformation of ent-trachylobane skeleton into ent-kaur-11-ene derivatives, Fraga and co-workers described the microbiological transformation of trachinodiol 77 by the fungus Mucor plumbeus,82 which led five polar compounds 78–82 (Scheme 13). The biotransformation of trachinodiol 77 to give sicanatriol 81 must occur via enzymatic abstraction of an H-11, this abstract is favoured because such carbon is allylic to the cyclopropane ring, with concomitant cleavage of this ring, giving a carbenium ion at C-16, subsequently neutralized with an OH anion, probably of water origin, to form the alcohol 81 (Scheme 14).
 | ||
| Scheme 13 Biotransformation of trachinodiol 77 by the fungus M. plumbeus. | ||
 | ||
| Scheme 14 Mechanism of biotransformation of trachinodiol 77 into alcohol 81. | ||
Litaudon and co-workers screened a range of oxidizing fungi (Rhizopus arrhizus, Aspergillus terreus, Beauveria bassiana, Mucor plumbeus, and Cylindrocarpon radicicola) for the biotransformation of ent-trachyloban-18-oic acid 83.83 In this case, biotransformation of 83 by Rhizopus arrhizus (this fungus was selected because it had the highest bioconversion yield and the highest diversity of metabolites) gave six oxidized compounds (Scheme 15), four of them rearranged derivatives by cleavage of the cyclopropane ring. Compounds 84 and 85 resulted from a direct enzymatic hydroxylation of compound 83 at positions C-17 and C-1, respectively, whereas a backbone rearrangement prior to the oxidation of ent-trachyloban-18-oic acid 83 occurred for compounds 87–89. Finally, compound 86, was a rearranged product.
 | ||
| Scheme 15 Biotransformation of ent-trachyloban-18-oic acid 83 by the fungus R. arrhizus. | ||
The biotransformation experiment of trachyloban-19-oic acid 90, epimeric compound of 83, carried out with R. stolonifer,84 yielded two trachylobane type compounds (Scheme 16), the C-7β 91 and the C-17 92 hydroxyl derivatives, and two rearranged ent-kaur-11-en-19-oic acids, the 16α 93 and the 9β,16α 94 hydroxylated compounds.
 | ||
| Scheme 16 Biotransformation of trachyloban-19-oic acid 90 by the fungus R. stolonifer. | ||
From a biosynthetic point of view, it is interesting to note that kaur-11-ene derivatives are scarce in nature and have only been found in plants where diterpenes with a trachylobane skeleton have also been isolated. Moreover, the rearrangement of a trachylobane diterpene lead ent-kaur-11-ene derivatives described below. All this facts support the biogenetic proposal based on the mechanism proposed by Fraga and co-workers, similar to that postulated by authors in plants of the Sideritis genus.81d,85
6. Kaurenes
Kaurenes represent an important group of tetracyclic diterpenes. Their structures are constituted by a perhydrophenanthrene unit (A, B and C rings) fused with a cyclopentane unit (D ring) formed by a bridge of two carbons between C-8 and C-13. ent-Kaurenes and many natural derivatives of these diterpenes have significant anti-inflammatory, anti-hypertensive, and diuretic effects in vivo, in addition to antimicrobial, smooth muscle relaxant, and cytotoxic actions in vitro.86Kauren-19-oic acid is one of the intermediate compounds involved in the biosynthesis of diverse kaurene diterpenes, including gibberellins, which represent an important group of growth phytohormones. Moreover, Kaurenoic acid is a bioactive compound with proven anticonvulsant, sedative, and hypotensive effects.87 Microbiological transformations of diterpenoids having ent-kaurane skeletons have been widely investigated, leading to a large variety of functionalized compounds.
The microbiological transformation of candidiol (15α,18-dihydroxy-ent-kaur-16-ene) 95a and 15α,19-dihydroxy-ent-kaur-16-ene 95b by Mucor plumbeus yielded the same hydroxylations (at C-3α, C-6α or C-11β) and epoxidation of the exocyclic double bond reactions (Scheme 17).88 This means that a change in the spatial orientation of the hydroxymethylene group at C-4, from equatorial in 95a to axial in 95b, does not affect the way in which these ent-kaurenes bind to the oxidative enzymes. Glucosides were formed in the feeding of 95b (α-axial CH2OH), but not in that of 95a (β-equatorial CH2OH). This was the first time that ent-kaurene derivatives of this type are formed in a biotransformation by a Mucor species. Other glucosyl derivatives had been obtained in the feeding of the mycotoxin zearalenone89 and the steroid resibufogenin90 with other species of Mucor.
 | ||
| Scheme 17 Microbiological transformation of 95a and 95b by Mucor plumbeus. | ||
In the case of candicandiol (7α,18-dihydroxy-ent-kaur-16-ene) 105 and epicandicandiol (7β,18-dihydroxy-ent-kaur-16-ene) 106,91 which differs both in the spatial change and in the orientation of the 7-hydroxyl group from equatorial in 105 to axial in the substrate 106. The main difference in its fermentation with Mucor plumbeus was the formation of a 3α 107 and 9β 110 hydroxylated derivative (Scheme 18), respectively, therefore a spatial change in the orientation of the hydroxyl group at C-7 affected the way in which these kaurenes bind to the oxidative enzymes affording a different hydroxylation pattern in the A and B rings. The formation of 16,17-dihydroxylated compounds, 109 and 112, can be explained by enzymatic epoxidation of the exocyclic double bond to give the corresponding epoxides, followed by opening of these in the medium. For the formation of sideritriol 108 and of canditriol 111, the authors proposed a mechanism where the enzymatic abstraction of a hydrogen at C-15 in with formation of a carbonium ion, followed by migration of the double bond to the 15,16-position and neutralization of the cation at C-17 by a hydroxyl group. Although these products could be biosynthesized by enzymatic opening of the 15,16-epoxide, this second mechanism is discarded, because analogously in the biotransformation of candicandiol 105, the 7α-epimer of the diterpene sideritriol 108, was not isolated.
 | ||
| Scheme 18 Microbiological transformation of 105 and 106. | ||
To evaluate the importance of the presence of a 15β-alcohol and a 3-oxo group in the biotransformation of ent-kaurene diterpenes by the fungus Gibberella fujikuroi, Fraga and co-workers prepared substrates 113 and 114 from 3α-hydroxy-15β-angeloxy-ent-kaur-16-ene92 previously isolated from Elaseolinum tenuifolium.93 The biotransformations with the fungus G. fujikuroi (Scheme 19) in the presence of AMO 1618, a compound that inhibits the production of ent-kaur-16-ene without affecting the post-kaurene metabolism,94 yielded the oxidation at C-19, 122–123, so the existence of a 15β-hydroxyl group in the molecule does not inhibit this functionalization, in contrast to previous studies with the presence of 15α-alcohol,95 and directs the hydroxylation at C-11β 115–121 and C-7α 116, 119–121, and 124–125. The occurrence of 11β-hydroxylated ent-kaur-16-ene diterpenes with oxygen functions of the 15α-OH, 15β-OH, or 15-oxo type in liverworts96 and higher plants97 seems to indicate that the 11β-hydroxylation should be directed by the presence in the molecule of a 15-oxygenated substituent, as occurs in the microbiological transformations with the fungus G. fujikuroi. A 3-oxo group in the molecule inhibits oxidation at C-19 115–119, but the presence of a 2,3-double bond does not affect this oxidation 122–123.
 | ||
| Scheme 19 Microbiological transformation of 107 and 108 by G. fujikuroi. | ||
7. Beyeranes
Stevioside, the major constituent of Stevia rebaudiana leaves extract (family Asteraceae), consists of a glycoside possessing steviol, an ent-beyerane tetracyclic diterpene with a ketone on the D ring, as its aglycon.98 Stevioside is approximately 300 times sweeter than sucrose and is widely used as a noncaloric sugar substitute in many kinds of foods and as a food supplement in many countries of Asia and South America.99 In addition to their sweetening properties, stevioside and steviol have been used to treat metabolic syndromes, such as hypertension, hyperglycemia, dyslipidemia, and diabetes. Its analog, isosteviol, produces vasodilation of rat aorta.100 Isosteviol lactone 169 has been used in biotransformations, because small modifications to the structure of a compound can modify its biological activities.101Many microorganisms are capable of reproducibly converting isosteviol 127 into many metabolites. To measure the antihypertensive activity of hydroxylated isosteviol, Liu and co-workers studied the microbial transformations of 127, obtained by hydrolysis of stevioside 126 with dilute hydrochloric acid, by Cunninghamella bainieri, Actinoplanes sp., Mucor recurvatus, and Cunninghamella blakesleeana (Scheme 20).102 The specie of C. bainieri have the ability to functionalize isosteviol 127 at the C-7 position, this is a common occurrence for other microbes with kaurane and beyerane skeletons,92,103 while hydroxylation at the 9β-position is obtained for the first time on a beyerane skeleton. Previously, the dihydroxylation on isosteviol 127 was reported only at the 1α- and 7β-positions,103b but in this case, several hydroxylated isosteviol analogues were prepared by introduction of a hydroxyl group at: 11β,12β 132 and 11β,12β,17 133 by Actinoplanes sp.; 12β,15β 130 and 7β,15β 131 by Mucor revurvatus; 7β 128 and 7α 129 by Cunninghamella bainieri; 7β 128, 9β 134 and 12β 135 by Cunninghamella blakesleeana.
 | ||
| Scheme 20 Microbiological transformation of isosteviol 127. | ||
In a posterior work, the same authors used three filamentous fungi, Mucor recurvatus (MR 36), Absidia pseudocylindrospora (ATCC 24169), and Aspergillus niger (BCRC 32720), for the biotransformation of 127 (Table 1).104 The reactions involved regio- and stereoselectively introduction of hydroxyl groups at A, B, C, and D rings of isosteviol 127. A comparison of the substrate specificity between these three selected fungi suggests that they possess different characteristics of reaction selectivity. M. recuvatus performs only mono and dihydroxylation on 127; A. pseudocylindrospora is able to specifically hydroxylate 127 at C-17; while, A. niger has the ability to dihydroxylate 127 at the 1α,7β- and 7β,11β-positions followed by oxidation of the 1α- or 7β-OH, respectively, to yield the metabolites 151–153. The dihydroxylation that occurred at the 1α,7β-positions of 127 by A. niger has previously been reported,103b although not the subsequent oxidation of an OH group to a ketone group.
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Substrate | Product | Organism | Ref. | ||||||||||
| R1 | R2 | R3 | R4 | R5 | R6 | R7 | R8 | R9 | R10 | ||||
| Isosteviol 127 | 137 | H | H | H | β-OH | H | H | H | CH3 | H | H | M. recurvatus | 104 |
| A. niger | 106 | ||||||||||||
| Isosteviol 127 | 138 | H | H | H | α-OH | H | H | H | CH3 | H | H | M. recurvatus | 104 |
| Isosteviol 127 | 139 | H | H | H | H | H | H | H | CH3 | OH | H | M. recurvatus | 104 |
| Isosteviol 127 | 140 | H | H | H | H | H | H | β-OH | CH3 | OH | H | M. recurvatus | 104 |
| Isosteviol 127 | 141 | H | H | H | β-OH | H | H | H | CH3 | OH | H | M. recurvatus | 104 |
| Isosteviol 127 | 142 | H | H | H | H | OH | H | H | CH2OH | H | H | A. pseudocylindrospora | 104 |
| Steviol-16α,17-epoxide 136 | C. bainieri | 105 | |||||||||||
| Isosteviol 127 | 143 | H | H | H | H | H | H | β-OH | CH2OH | H | H | A. pseudocylindrospora | 104 |
| Steviol-16α,17-epoxide 136 | S. griseus | 105 | |||||||||||
| Isosteviol 127 | 144 | H | H | H | H | H | H | H | α-OH | CH2OH | H | A. pseudocylindrospora | 104 |
| Steviol-16α,17-epoxide 136 | S. griseus | 105 | |||||||||||
| Isosteviol 127 | 145 | H | H | H | α-OH | H | H | H | CH2OH | H | H | A. pseudocylindrospora | 104 |
| Steviol-16α,17-epoxide 136 | C. bainieri | 105 | |||||||||||
| Isosteviol 127 | 146 | H | H | H | α-OH | H | H | β-OH | CH3 | H | H | A. pseudocylindrospora | 104 |
| Isosteviol 127 | 147 | α-OH | H | H | H | H | H | H | CH3 | H | H | A. niger | 104 |
| Isosteviol 127 | 148 | α-OH | H | H | β-OH | H | H | H | CH3 | H | H | A. niger | 104 |
| Isosteviol 127 | 149 | H | H | H | ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
H | OH | H | CH3 | H | H | A. niger | 104 |
| Isosteviol 127 | 150 | α−OH | H | H | O |
H | H | H | CH3 | H | H | A. niger | 104 |
| Isosteviol 127 | 151 | O |
H | H | β-OH | H | H | H | CH3 | H | H | A. niger | 104 |
| Isosteviol 127 | 152 | H | H | H | β-OH | H | OH | H | CH3 | H | H | A. niger | 104 |
| Isosteviol 127 | 153 | α−OH | H | H | β-OH | H | H | H | CH3 | H | H | A. niger | 104 |
| Isosteviol 127 | 154 | α−OH | H | OH | β-OH | H | H | H | CH3 | H | H | A. niger | 104 |
| Isosteviol 127 | 155 | H | H | H | H | H | OH | H | CH3 | H | H | A. niger | 106 |
| Isosteviol 127 | 156 | H | H | H | H | H | H | β-OH | CH3 | H | H | A. niger | 106 |
| Isosteviol 127 | 157 | H | H | H | H | H | H | H | CH2OH | H | H | G. cingulata | 106 |
| Steviol-16α,17-epoxide 136 | S. griseus | 105 | |||||||||||
| Isosteviol 127 | 158 | H | H | H | O |
H | H | H | CH3 | H | H | M. elongate | 106 |
| Steviol-16α,17-epoxide 136 | 159 | H | OH | H | H | H | H | H | CH2OH | H | H | S. griseus | 105 |
| Steviol-16α,17-epoxide 136 | 160 | H | H | H | H | H | H | H | CH2OH | H | OH | S. griseus | 105 |
| Steviol-16α,17-epoxide 136 | 161 | OH | H | H | H | H | H | H | CH2OH | H | H | C. bainieri | 105 |
| Steviol-16α,17-epoxide 136 | 162 | H | H | H | β-OH | H | H | H | CH2OH | H | H | C. bainieri | 105 |

Metabolites 142–145 were obtained previously from incubation of steviol-16α,17-epoxide 136, prepared from isosteviol 127 by a reaction with m-chloroperbenzoic acid, with Streptomyces griseus and Cunninghamella bainieri (Table 1).105 These two microorganisms have the abilities to produce not only regio- and stereoselective hydroxylation but also to rearrange the ent-kaurane into an ent-beyerane skeleton. While Akihisa and co-workers106 obtained 7β-hydroxyisosteviol 137, together with 11β-hydroxyisosteviol 155 and 12β-hydroxyisosteviol 156, from microbial transformation of isosteviol 127 by the fungus Aspergillus niger; 17-hydroxyisosteviol 157 by the fungus Glomerella cingulata; and 7-oxoisosteviol 158 by the fungus Mortierella elongate (Table 1).
Yang, Lin and co-workers, studied the microbial biotransformation of isosteviol oxime (ent-16-E-hydroxyiminobeyeran-19-oic acid) 163, prepared by reacting 127 with hydroxylamine hydrochloride, by A. niger and A. pseudocylindrospora,107 which performed hydroxylation reactions 164–167, Beckmann rearrangement 171, and an abnormal Beckmann rearrangement 169–170 (Scheme 21). Although the isoteviol lactone 168 has been prepared previously by chemical methods, with acid catalysis (concentrated hydrochloric acid or 25% sulphuric acid) in an ampoule at 180 °C,108 this biotransformation yield this product in mild conditions, although in lower yields. The authors propose two possible mechanisms for the formation of isoteviol lactone 168. According to the literature,108 nitrile carbocation A (Scheme 22) is formed as in the Beckmann fragmentation reaction, however, it is further stabilised not by losing α-proton but by attaching a hydroxyl group and subsequent cyclization in imidate B, which is unstable and easily hydrolyses to lactone 168. On the other hand, the isolation of ketone 168, also suggests that substrate 163 might be hydrolyzed to isosteviol 127 first, and then converted to the 167 and 168 by hydroxylation and Baeyer–Villiger reaction, as reported in a previous works by Fraga and co-workers.30,31
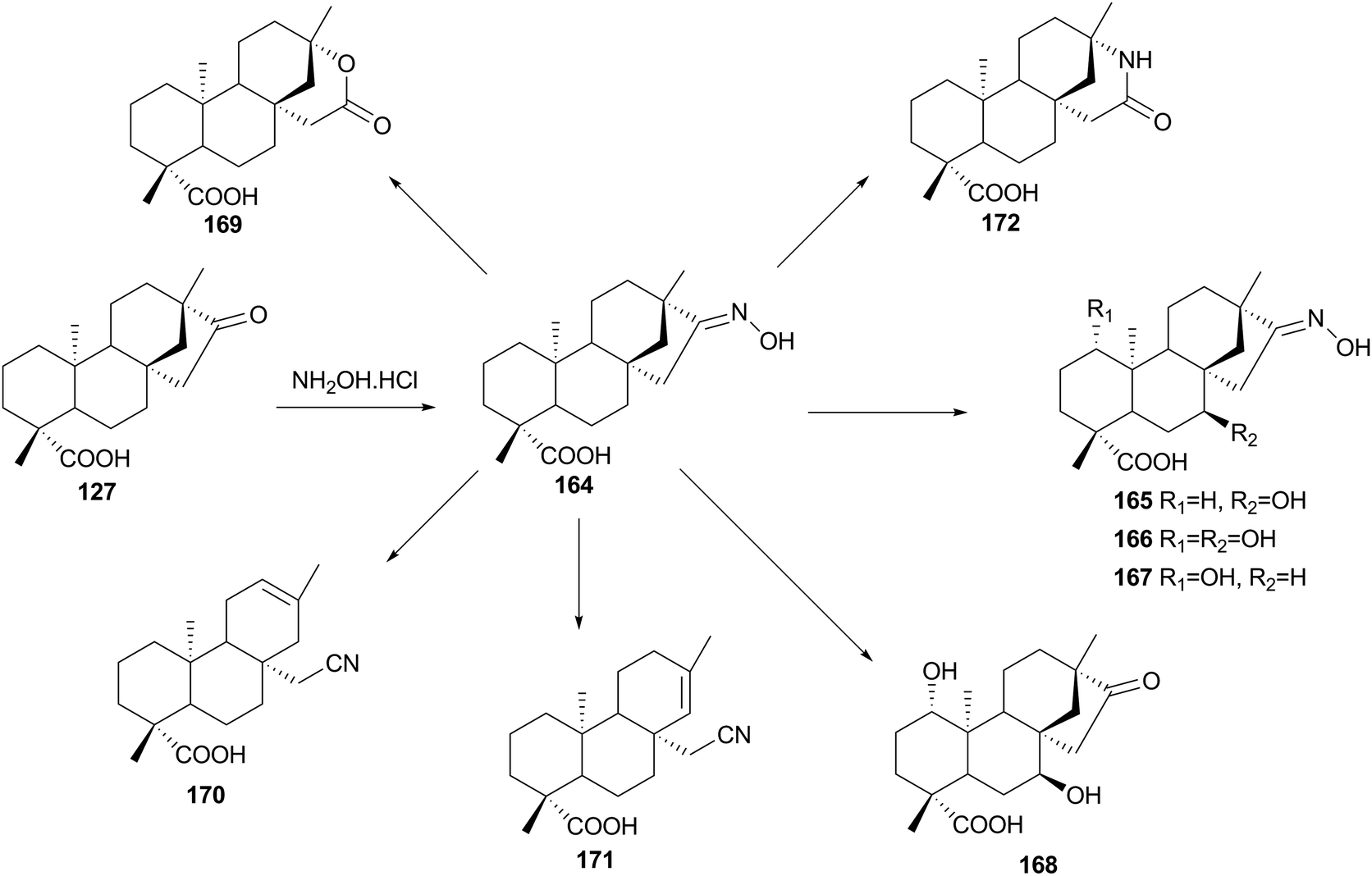 | ||
| Scheme 21 Biotransformation of isosteviol 16-oxime 164 by Aspergillus niger and Absidia pseudocylindrospora. | ||
 | ||
| Scheme 22 Possible mechanism for isoteviol lactone 169 formation. | ||
Twenty-five selected microbial cultures were used to identify organisms capable of metabolizing isoteviol lactone 168,109 from these, Cunninghamella bainieri (ATCC 9244) and Aspergillus niger (BCRC 32720) were selected as biocatalysts for scaled-up biotransformations. Incubation of 168 with C. bainieri (Table 2) afforded metabolites which involved isomerisation of lactone moiety 178–179, hydroxylation 173 and 179, and ring cleavage reactions followed by oxidation and selective O-methylation 182. While A. niger possesses the abilities to not only isomerize the lactone ring to 178, but to cause regio- and stereoselective mono-, di-, and trihydroxylation at the 1α-, 7β-, and 11β-positions.
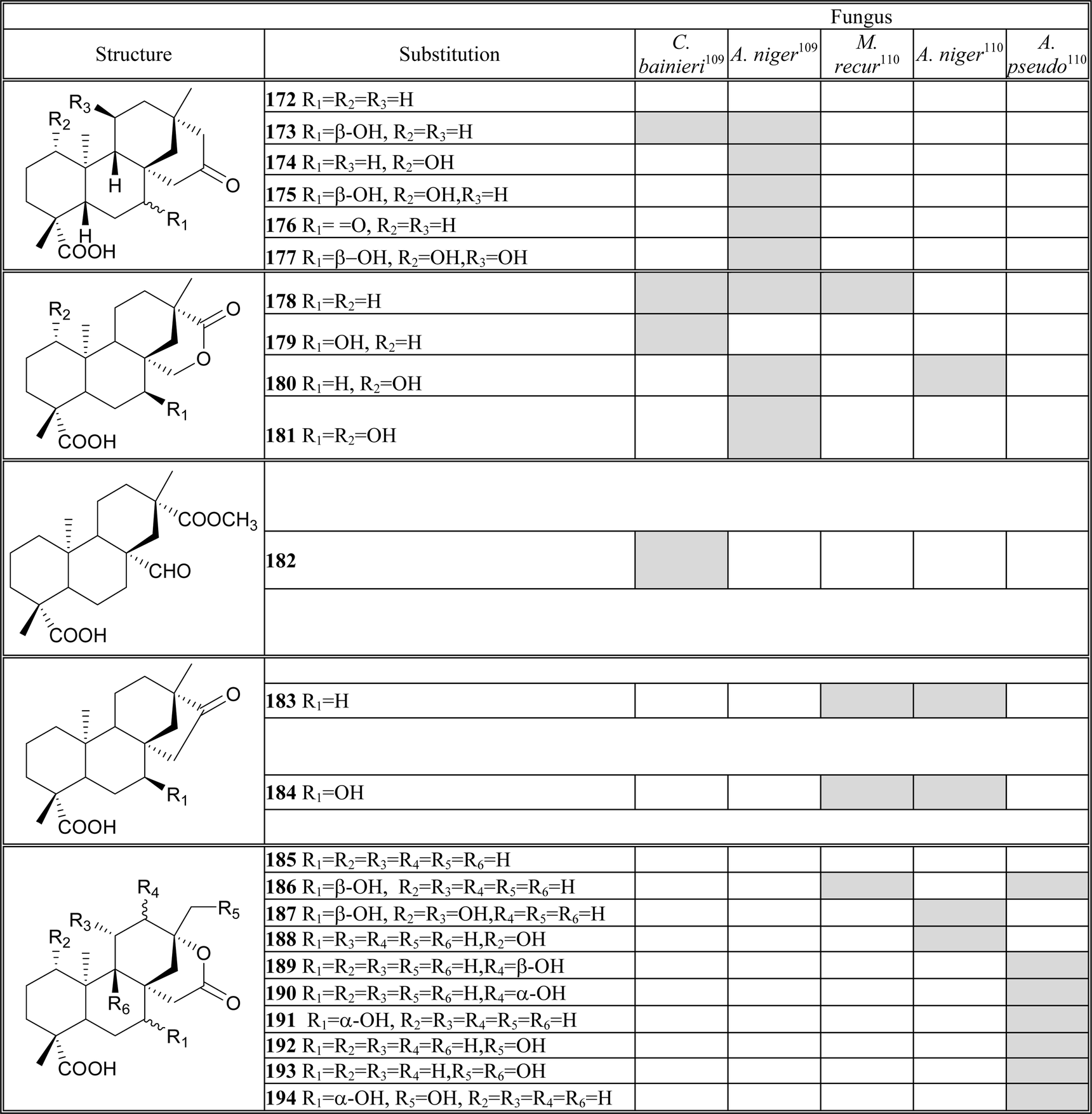 |
The change of fungi to Mucor recurvatus (MR 36), Aspergillus niger (BCRC 31130) and Absidia pseudocylindrospora (ATCC 24169), the same authors obtained in the incubation of 168 thirteen diterpenoids (Table 2),110 where 183–194 were different to the previous work.109 The reactions involved hydroxylation at the C-1, C-7, C-11, C-12 and C-17, and reduction to the ketone moiety 183–184.
8. Stemodanes
This type of compounds, isolated for the first time from Stemodia maritime L. (Scrophulariaceae) and used as a caribbean folk medicine for venereal diseases treatment,111 have a unique tetracyclic framework: a trans-decalin (ring A/B) fused to a bicyclo[3.2.1]octane (ring C/D). Stemodanes represent a synthetic challenge, and for this reason, structural modifications of these substrates and their derivatives via microbial transformation could be an alternative to obtain unique analogues with potential biological activity.Reese and co-workers incubated stemodin 195 and their dimethylcarbamate derivative 196 by means of cultures of the fungi Cunninghamella echinulata var. elegans (ATCC 8688a) and Phanerochaete chrysosporium (ATCC 24725) (Scheme 23).112 The protection of alcohols as their carbamates promote stronger interactions between the substrate and the hydrophobic amino acids that are believed to constitute the enzyme's active site, for the nonpolar nature of the group. Furthermore, it is proposed that an electro-negative oxygen atom would help to anchor the substrate there. Carbamate derivatives of various substrates have been used successfully to improve the bioconversion yields as well as varying the sites where hydroxylation occurs.23a,113
 | ||
| Scheme 23 Biotransformation of stemodin 195 and its carbamate derivative 196. | ||
Incubation of C. echinulata with stemodin 195 gave three trihydrolylated products 197–199 (Scheme 23), while the fermentation of 2α-(N,N-dimethylcarbamoxy)-13-hydroxystemodane 196 yielded only two monohydroxylated metabolites 201–202. On the other hand, P. chrysosporium converted stemodin 196 into 198–200. The dimethylcarbamate 196 was not transformed by this microorganism.
9. Conclusions
Biotransformation has gained significant importance in modifying the naturally occurring substance. In this review, we have highlighted the use of a series of diterpenes as starting materials for diverse biocatalytic strategies, in an effort to establish attractive methodologies for obtaining new pharmaceuticals, intermediates, and analytical reagents. Most biocatalytic reactions can be carried out under certain safety, health, environmental, and economical conditions (Table 3).| Microorganism | Substrate | Time/days | Number products | Modifications |
|---|---|---|---|---|
| A. pseudocylindrospora | ||||
| ATCC 24169 | Isosteviol | 6 | 6 | Hydroxylation |
| Isosteviol 16-E-oxime | 6 | 3 | Hydroxylation; hydrolysis oxime | |
| Isoteviol lactone | 6 | 7 | Hydroxylation | |
| Actinoplanes sp. | ||||
| Isosteviol | 6 | 2 | Hydroxylation | |
| A. niger | ||||
| BCRC 32720 | Isosteviol | 6 | 8 | Hydroxylation |
| Isosteviol | 7 | 3 | Hydroxylation | |
| Isosteviol 16-E-oxime | 6 | 8 | Hydroxylation; hydrolysis oxime; Beckmann and abnormal Beckmann rearrangement | |
| BCRC 32720 | Isoteviol lactone | 6 | 8 | Isomerization; hydroxylation |
| BCRC 31130 | Isoteviol lactone | 6 | 5 | Hydroxylation; reduction |
| A. ochraceus | ||||
| ent-8(14),15-Pimaradiene | 5 | 5 | Hydroxylation | |
| C. bainieri | ||||
| Isosteviol | 6 | 2 | Hydroxylation (C-7) | |
| Steviol-16α,17-epoxide | 6 | 6 | Oxidation; backbone rearrangement | |
| Isoteviol lactone | 6 | 4 | Isomerization; hydroxylation; ring cleavage reactions | |
| C. blakesleeana | ||||
| Isosteviol | 6 | 3 | Hydroxylation | |
| C. echinulata | ||||
| Stemodin | 10 | 3 | Hydroxylation | |
| Stemodin dimethylcarbamate derivative | 10 | 2 | Hydroxylation | |
| C. elegans | ||||
| Cryptotanshinone | 5 | 3 | Hydroxylation | |
| G. fujikuroi | ||||
| 2α,19-Dihydroxy-9-epi-ent-pimara-7,15-diene | 6 | 7 | Hydroxylation | |
| 18-Hydroxy-9-epi-ent-pimara-7,15-diene | 6 | 5 | Hydroxylation | |
| 19-Hydroxy-13-epi-ent-pimara-9(11),15-diene | 6 | 4 | Hydroxylation (C-2); oxidation (C1); Baeyer–Villiger reaction | |
| 13-epi-ent-Pimara-9(11),15-diene-19-oic acid | 6 | 4 | Epoxidation; hydroxylation (C-8); oxidation (C-7) | |
| 9,13-epi-ent-pimara-7,15-diene diterpene | 1 | 6 | Hydroxylation, epoxidation | |
| 15α-Hydroxy-3-oxo-ent-kaur-16-ene | 6 | 5 | Hydroxylation; oxidation (C-19) | |
| 15α-Hydroxy-ent-kaur-2,16-diene | 6 | 6 | Hydroxylation; oxidation (C-19) | |
| G. cingulata | ||||
| ent-Pimara-8(14),15-dien-19-oic acid | 7 | 1 | Reduction of acid moiety | |
| Isosteviol | 7 | 1 | Hydroxylation | |
| M. isabellina | ||||
| Dehydroabietic acid | 3 | 1 | Hydroxylation (C-2α) | |
| M. elongate | ||||
| Isosteviol | 7 | 2 | Oxidation | |
| M. circinelloides | ||||
| Dehydroabietic acid | 3 | 1 | Hydroxylation (C-2α) | |
| M. plumbeus | ||||
| Dehydroabietanol | 6 | 6 | Hydroxylation | |
| Teideadiol | 6 | 3 | Hydroxylation | |
| Trachinodiol | 6 | 5 | Hydroxylation; backbone rearrangement | |
| Candidiol | 6 | 5 | Hydroxylation | |
| 15α,19-Dihydroxy-ent-kaur-16-ene | 6 | 7 | Hydroxylation | |
| Candicandiol | 6 | 3 | Hydroxylation | |
| Epicandicandiol | 6 | 3 | Hydroxylation | |
| M. recurvatus | ||||
| Isosteviol | 6 | 2 | Hydroxylation | |
| (MR 36) | Isosteviol | 6 | 5 | Hydroxylation |
| Isoteviol lactone | 6 | 4 | Hydroxylation reduction | |
| M. rouxii | ||||
| ent-Pimara-8(14),15-dien-19-oic acid | 7 | 2 | Isomerization double bond; oxidation (C-7) | |
| Nocardia sp. NRRL 5646 | ||||
| Carnosic acid | 3 | 3 | Hydroxylation; methylation | |
| P. chrysosporium | ||||
| ATCC 24725 | Stemodin | 10 | 3 | Hydroxylation |
| Stemodin dimethylcarbamate derivative | 10 | 0 | ||
| R. arrhizus | ||||
| ent-Trachyloban-18-oic acid | 14 | 6 | Hydroxylation; backbone rearrangement | |
| R. stolonifer | ||||
| Clerodane lactone | 7 | 2 | Oxidation furan; allylic hydroxylation | |
| Clerodane methyl ester | 7 | 4 | Oxidation furan | |
| Trachyloban-19-oic acid | 20 | 4 | Hydroxylation; backbone rearrangement | |
| S. griseus | ||||
| ATCC 10137 | Steviol-16α,17-epoxide | 6 | 7 | Hydroxylation; oxidation; backbone rearrangement |
The main structural modification observed is the regiospecific and stereospecific hydroxylation, and the position on the molecule where this occurs depends on both the substrate and the microorganism. Among the most frequently used microorganisms are Mucor plumbeus, Gibberella fujikuroi, and Aspergillus niger (Fig. 1). The number of products resulting from biotransformation seem to vary with the microorganism, with few exceptions, microbial metabolism usually results in the generation of multiple products, which complicates downstream processing, in customarily low yields.
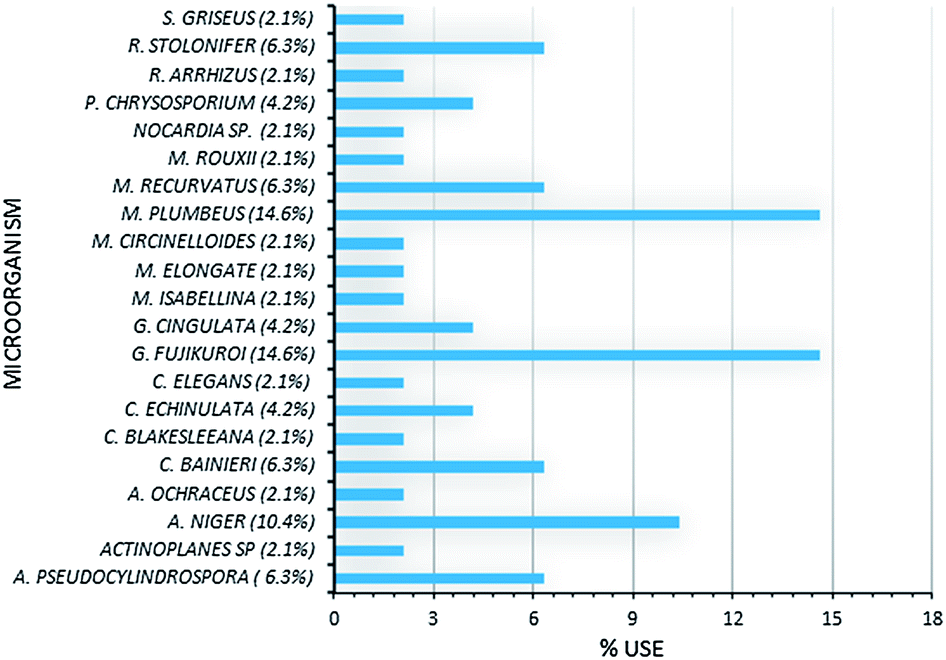 | ||
| Fig. 1 Comparative involvement of microorganisms utilized for biotransformation. | ||
Microbial biotransformation procedures documented here can be used to obtain diterpenoid derivatives hydroxylated in almost any position. These derivatives are of greatest interest because they increase solubility and provide sites for further modification. Future research should include the search of additional fungal and bacterial strains, the use of biocatalytic reactions on solid-supported substrates, as well as the integration of biocatalysis and organic synthesis to the creation of new synthetic strategies.
Acknowledgements
This work was economically supported by SIP-IPN. MSOR and JGM are also grateful for the grants from SNI-CONACyT. MRM acknowledge the PIFI-IPN scholarship.References
- (a) J. Hong-Fang, L. Xue-Juan and Z. Hong-Yu, EMBO Rep., 2009, 10, 194 CrossRef PubMed; (b) D. J. Newmann, M. C. Gordon and K. M. Snader, J. Nat. Prod., 2003, 66, 1022 CrossRef PubMed.
- (a) J. C. Sacchettini and C. D. Poulter, Science, 1997, 277, 1788 CrossRef CAS; (b) T. J. Maimone and P. Baran, Nat. Chem. Biol., 2007, 3, 396 CrossRef CAS PubMed.
- M. Rodriguez-Concepcion and A. Boronat, Plant Physiol., 2002, 130, 1079 CrossRef CAS PubMed.
- (a) T. Rabi and A. Bishayee, Breast Cancer Res. Treat., 2009, 115, 223 CrossRef CAS PubMed; (b) K. H. Wagner and I. Elmadfa, Ann. Nutr. Metab., 2003, 47, 95 CrossRef CAS PubMed; (c) N. Sultana and A. Ata, J. Enzyme Inhib. Med. Chem., 2008, 23, 739 CrossRef CAS PubMed; (d) B. A. Shah, G. N. Qazi and S. C. Taneja, Nat. Prod. Rep., 2009, 26, 72 RSC.
- (a) S. Aggarwal, Y. Takada, S. Singh, J. Myers and B. Aggarwal, Int. J. Cancer, 2004, 111, 679 CrossRef CAS PubMed; (b) J. Molnar, N. Gyémánt, M. Tanaka, J. Hohmann, E. Bergmann-Leitner, P. Molnár, J. Deli, R. Didiziapetris and M. J. Ferreira, Curr. Pharm. Des., 2006, 12, 287 CrossRef CAS.
- B. de las Heras, B. Rodriguez, L. Bosca and A. M. Villar, Curr. Top. Med. Chem., 2003, 3, 171 CrossRef CAS.
- J. S. Dickschat, Nat. Prod. Rep., 2011, 28, 1917 RSC.
- V. E. J. Hikawczuk, J. R. Saad, O. S. Giordano, C. García, T. Martín, V. S. Martín, M. E. Sosa and C. E. Tonn, J. Nat. Prod., 2008, 71, 190 CrossRef PubMed.
- (a) C. V. Gómez, M. Martínez-Vázquez and B. Esquivel, Z. Naturforsch., C: J. Biosci., 2009, 64, 502 Search PubMed; (b) E. A. K. Gebbinck, B. J. M. Jansen and A. Groot, Phytochemistry, 2002, 61, 737 CrossRef.
- (a) B. M. Fraga, C. E. Díaz, A. Guadaño and A. González-Coloma, J. Agric. Food Chem., 2005, 53, 5200 CrossRef CAS PubMed; (b) G. B. Messiano, L. Vieira, M. B. Machado, L. M. Lopes, S. A. de Bortoli and J. Zukerman-Schpector, J. Agric. Food Chem., 2008, 56, 2655 CrossRef CAS PubMed.
- J. Becerra, C. Flores, J. Mena, P. Aqueveque, J. Alarcon, M. Bittner, V. Hernández, M. Hoeneisen, E. Ruiz and M. Silva, J. Chil. Chem. Soc., 2002, 47, 151 CAS.
- (a) J. R. Hanson, Nat. Prod. Rep., 2011, 28, 1755 RSC; (b) R. J. Peters, Nat. Prod. Rep., 2010, 27, 1521 RSC.
- A. F. Barrero, M. M. Herrador, P. Arteaga, J. F. Arteaga and A. F. Arteaga, Molecules, 2012, 17, 1448 CrossRef CAS PubMed.
- T. Yosief, A. Rudi, Z. Stein, I. Goldberg, G. M. D. Gravalos, M. Schleyer and Y. Kashman, Tetrahedron Lett., 1998, 39, 3323 CrossRef CAS.
- X. S. Pan, M. Dias, M. Palumbo and L. M. Fisher, Nucleic Acids Res., 2008, 36, 5516 CrossRef CAS PubMed.
- A. Ortega, J. F. Blount and P. S. Manchand, J. Chem. Soc., Perkin Trans. 1, 1982, 11, 2505 RSC.
- B. E. Kane, C. R. McCurdy and D. M. Ferguson, J. Med. Chem., 2008, 51, 1824 CrossRef CAS PubMed.
- S. Malik, R. M. Cusidó, M. H. Mirjalili, E. Moyano, J. Palazón and M. Bonfill, Process Biochem., 2011, 46, 23 CrossRef CAS PubMed.
- (a) P. Vuorela, M. Leinonen, P. Saikku, P. Tammela, J. P. Rauha, T. Wennberg and H. Vuorela, Curr. Med. Chem., 2004, 11, 1375 CrossRef CAS; (b) D. J. Newman, J. Med. Chem., 2008, 51, 2589 CrossRef CAS PubMed.
- (a) S. R. Ambrosio, C. R. Tirapelli, F. B. Da Costa and A. M. De Oliveira, Life Sci., 2006, 79, 925 CrossRef CAS PubMed; (b) C. R. Tirapelli, S. R. Ambrosio, F. B. Da Costa and A. M. De Oliveira, Recent Pat. Cardiovasc. Drug Discovery, 2008, 3, 1 CAS.
- (a) W. A. Duetz, J. B. Van Beilen and B. Witholt, Curr. Opin. Biotechnol., 2001, 12, 419 CrossRef CAS; (b) S. F. Arantes and J. R. Hanson, Curr. Org. Chem., 2007, 11, 657 CrossRef CAS.
- H. F. de Castro and W. A. Anderson, Quim. Nova, 1995, 18, 544 CAS.
- (a) G. O. Buchanan and P. B. Reese, Phytochemistry, 2001, 56, 141 CrossRef CAS , and references therein; (b) W. Gladkowski, M. Grabarczyk, K. Winska, B. Ratus, A. Bialonska, Z. Ciunik and C. Wawrzenczyk, J. Mol. Catal. B: Enzym., 2007, 49, 79 CrossRef CAS PubMed; (c) R. K. Venisetty and V. Ciddi, Curr. Pharm. Biotechnol., 2003, 4, 153 CrossRef CAS.
- H. L. Holland and H. K. Weber, Curr. Opin. Biotechnol., 2000, 11, 547 CrossRef CAS.
- L. R. Lehman and J. D. Stewart, Curr. Org. Chem., 2001, 5, 439 CrossRef CAS.
- (a) A. T. Merritt and S. V. Ley, Nat. Prod. Rep., 1992, 9, 243 RSC; (b) A. Castro and J. Coll, Nat. Prod. Commun., 2008, 3, 1021 CAS; (c) Y. B. Wu, Z. Y. Ni, Q. W. Shi, M. Dong, H. Kiyota, Y. C. Gu and B. Cong, Chem. Rev., 2012, 112, 5967 CrossRef CAS PubMed.
- T. Tokoroyama, Synthesis, 2000, 611 CrossRef CAS PubMed.
- M. I. Choudhary, M. Y. Mohammad, S. G. Musharraf, I. Onajobi, A. Mohammad, I. Anis, M. R. Shah and A. U. Rahman, Phytochemistry, 2013, 90, 56 CrossRef PubMed.
- L. A. Peterson, Chem. Res. Toxicol., 2013, 26, 6 CrossRef CAS PubMed.
- (a) B. M. Fraga, P. González, M. G. Hernández, M. C. Chamy and J. A. Garbarino, Phytochemistry, 1998, 47, 211 CrossRef CAS; (b) B. M. Fraga, M. G. Hernández, P. González, M. C. Chamy and J. A. Garbarino, Phytochemistry, 2000, 53, 395 CrossRef CAS.
- B. M. Fraga, R. Guillermo, M. G. Hernández, M. C. Chamy and J. A. Garbarino, J. Nat. Prod., 2009, 72, 87 CrossRef CAS PubMed.
- B. M. Fraga, P. González, M. G. Hernández and S. Suárez, Tetrahedron, 2005, 61, 5623 CrossRef CAS PubMed.
- A. Rioz-Martínez, G. de Gonzalo, D. E. T. Pazmiño, M. W. Fraaije and V. Gotor, Eur. J. Org. Chem., 2009, 2526 CrossRef.
- For some examples, see: (a) M. D. Mihovilovic, P. Kapitán and P. Kapitánová, ChemSusChem, 2008, 1, 143 CrossRef CAS PubMed; (b) P. Cernuchová and M. D. Mihovilovic, Org. Biomol. Chem., 2007, 5, 1715 RSC; (c) R. Snajdrova, G. Grogan and M. D. Mihovilovic, Bioorg. Med. Chem. Lett., 2006, 16, 4813 CrossRef CAS PubMed.
- S. R. Ambrosio, K. Schorr and F. B. Da Costa, Biochem. Syst. Ecol., 2004, 32, 221 CrossRef CAS.
- M. E. Severiano, M. R. Simao, T. S. Porto, C. H. G. Martins, R. C. S. Veneziani, N. A. J. C. Furtado, N. S. Arakawa, S. Said, D. C. R. de Oliveira, W. R. Cunha, L. E. Gregorio and S. R. Ambrosio, Molecules, 2010, 15, 8553 CrossRef CAS PubMed.
- T. S. Porto, M. R. Simão, L. Z. Carlos, C. H. G. Martins, N. A. J. C. Furtado, S. Said, N. S. Arakawa, R. A. dos Santos, R. C. S. Veneziani and S. R. Ambrósio, Phytother. Res., 2013, 27, 1502 CAS.
- B. M. Fraga, P. González, M. G. Hernández, M. C. Chamy and J. A. Garbarino, Phytochem. Lett., 2009, 2, 201 CrossRef CAS PubMed.
- (a) D. Martin, D. Tholl, J. Gershenzon and J. Bohlmann, Plant Physiol., 2002, 129, 1003 CrossRef CAS PubMed; (b) A. San Feliciano, M. Gordaliza, M. A. Salinero and J. M. Miguel del Corral, Planta Med., 1993, 59, 485 CrossRef CAS PubMed.
- For some examples, see: (a) B. Sepúlveda, L. Astudillo, J. A. Rodríguez, T. Yáñez, C. Theoduloz and G. Schmeda-Hirschmann, Pharmacol. Res., 2005, 52, 429 CrossRef PubMed; (b) B. Gigante, A. M. Silva, M. J. Marcelo-Curto, S. S. Feio, J. Roseiro and L. V. Reis, Planta Med., 2002, 68, 680 CrossRef CAS PubMed; (c) G. M. Woldemichael, G. Wachter, M. P. Singh, W. M. Maiese and B. N. Timmermann, J. Nat. Prod., 2003, 66, 242 CrossRef CAS PubMed; (d) W. Gu and S. F. Wang, Eur. J. Med. Chem., 2010, 45, 4692 CrossRef CAS PubMed; (e) Y. Kinouchi, H. Ohtsu, H. Tokuda, H. Nishino, S. Matsunaga and R. Tanaka, J. Nat. Prod., 2000, 63, 817 CrossRef CAS PubMed; (f) S. Prinz, U. Mullner, J. Heilmann, K. Winkelmann, O. Sticher, E. Haslinger and A. Hufner, J. Nat. Prod., 2002, 65, 1530 CrossRef CAS PubMed; (g) S. F. H. Zaidi, S. Awale, S. K. Kalauni, Y. Tezuka, H. Esumi and S. Kadota, Planta Med., 2006, 72, 1231 CAS; (h) X. Rao, Z. Song, L. He and W. Jia, Chem. Pharm. Bull., 2008, 56, 1575 CrossRef CAS.
- T. Ohwada, T. Nonomura, K. Maki, K. Sakamoto, S. Ohya, K. Muraki and Y. Imaizumi, Bioorg. Med. Chem. Lett., 2003, 13, 3971 CrossRef CAS PubMed.
- V. J. J. Martin and W. W. Mohn, J. Bacteriol., 2000, 182, 3784 CrossRef CAS.
- S. T. Häkkinen, P. Lackman, H. Nygrén, K. M. Oksman-Caldentey, H. Maaheimo and H. Rischer, J. Biotechnol., 2012, 157, 287 CrossRef PubMed.
- B. Schmidt, N. Joussen, M. Bode and I. Schuphan, Biochem. Soc. Trans., 2006, 34, 1241 CrossRef CAS PubMed.
- (a) V. J. J. Martin, Z. T. Yu and W. W. Mohn, Arch. Microbiol., 1999, 172, 131 CrossRef CAS; (b) S. N. Liss, P. A. Bicho and J. N. Saddler, Can. J. Microbiol., 1997, 43, 599 CrossRef CAS.
- (a) K. Mitsukura, T. Imoto, H. Nagaoka, T. Yoshida and T. Nagasawa, Biotechnol. Lett., 2005, 27, 1305 CrossRef CAS PubMed; (b) D. J. Smith, V. J. J. Martin and W. W. J. Mohn, J. Bacteriol., 2004, 186, 3631 CrossRef CAS PubMed.
- T. A. Van Beek, F. W. Claassen, J. Dorado, M. Godejohann, R. Sierra-Alvarez and J. B. P. A. Wijnberg, J. Nat. Prod., 2007, 70, 154 CrossRef CAS PubMed.
- A. A. Tapia, M. D. Vallejo, S. C. Gouiric, G. E. Feresin, P. C. Rossomando and D. A. Bustos, Phytochemistry, 1997, 46, 131 CrossRef CAS.
- S. C. Gouiric, G. E. Feresin, A. A. Tapia, P. C. Rossomando, G. Schmeda-Hirschmann and D. A. Bustos, World J. Microbiol. Biotechnol., 2004, 20, 281 CrossRef CAS.
- R. Ekman and R. Sjöholm, Acta Chem. Scand., Ser. B, 1979, 33, 76 CrossRef PubMed.
- (a) J. P. Kutney, M. Singh, G. M. Hewitt, P. J. Salisbury, B. R. Worth, J. A. Servizi, D. W. Martens and R. W. Gordon, Can. J. Chem., 1981, 59, 2334 CrossRef CAS; (b) J. P. Kutney, L. S. Choi, G. M. Hewitt, P. J. Salisbury and M. Singh, Appl. Environ. Microbiol., 1985, 49, 96 CAS.
- J. Biellman, G. Branlant, M. Gero-Robert and M. Poiret, Tetrahedron, 1973, 29, 1237 CrossRef.
- V. J. J. Martin and W. W. Mohn, J. Bacteriol., 1999, 181, 2675 CAS.
- J. F. Biellman, G. Branlant, M. Gero-Robert and M. Poiret, Tetrahedron, 1973, 29, 1227 CrossRef.
- A. Ulubelen and G. Topcu, Phytochemistry, 1992, 31, 3949 CrossRef CAS.
- (a) J. L. Breton, A. G. González and G. De Leon, An. Quim., 1970, 66, 293 Search PubMed; (b) B. M. Fraga, T. Mestres, C. E. Diaz and J. M. Arteaga, Phytochemistry, 1994, 35, 1509 CrossRef CAS.
- B. M. Fraga, M. G. Hernández, J. M. Artega and S. Suárez, Phytochemistry, 2003, 63, 663 CrossRef CAS.
- (a) A. González, L. San Andrés, Z. Aguiar and J. G. Luis, Phytochemistry, 1992, 31, 1297 CrossRef; (b) M. E. Curvelier, H. Richard and C. Berset, J. Am. Oil Chem. Soc., 1996, 73, 645 CrossRef; (c) S. S. Chang, B. Ostric-Matijaseuc, O. A. Hsieh and C. L. Huang, J. Food Sci., 1977, 42, 1102 CrossRef CAS.
- (a) E. Wenkert, A. Fuchs and J. D. McChesney, J. Org. Chem., 1965, 30, 2931 CrossRef CAS; (b) J. G. Luis, L. San Andrés and W. Q. Quiñones, Tetrahedron Lett., 1994, 35, 179 CrossRef CAS; (c) T. Masuda, Y. Inaba and Y. Takeda, J. Agric. Food Chem., 2001, 49, 5560 CrossRef CAS PubMed.
- (a) J. G. Marrero, L. San Andrés and J. G. Luis, J. Nat. Prod., 2002, 65, 986 CrossRef CAS PubMed; (b) J. G. Marrero, L. Moujir, L. San Andrés, N. P. Montaño, L. Araujo and J. G. Luis, J. Nat. Prod., 2009, 72, 1385 CrossRef CAS PubMed; (c) J. G. Marrero, L. San Andrés and J. G. Luis, J. Chem. Res., 2013, 37, 193 CrossRef CAS.
- J. Bauer, S. Kuehnl, J. M. Rollinger, O. Scherer, H. Northoff, H. Stuppner, O. Werz and A. Koeberle, J. Pharmacol. Exp. Ther., 2012, 342, 169 CrossRef CAS PubMed ; and the references cited therein.
- E. A. Offord, K. Mace, C. Ruffieux, A. Malone and A. M. Pfeifer, Carcinogenesis, 1995, 16, 2057 CrossRef CAS PubMed.
- K. W. Singletary, Cancer Lett., 1996, 100, 139 CrossRef CAS.
- M. Hosny, H. A. Johnson, A. K. Ueltschy and J. P. N. Rosazza, J. Nat. Prod., 2002, 65, 1266 CrossRef CAS PubMed.
- Y. Chen and J. P. N. Rosazza, Appl. Environ. Microbiol, 1994, 60, 1292 CAS.
- K. Dhar and J. N. P. Rosazza, Appl. Environ. Microbiol, 2000, 66, 4877 CrossRef CAS.
- X. Wang, S. L. Morris-Natschke and K. H. Lee, Med. Res. Rev., 2007, 27, 133 CrossRef CAS PubMed.
- J. W. Wang and J. Y. Wu, Appl. Microbiol. Biotechnol., 2010, 88, 437 CrossRef CAS PubMed.
- Y. Li, Y. Gong, L. Li, H. M. Abdolmaleky and J. R. Zhou, Mol. Carcinog., 2013, 52, 535 CrossRef CAS PubMed.
- R. Lin, W. R. Wang, J. T. Liu, G. D. Yang and C. J. Han, J. Ethnopharmacol., 2006, 108, 217 CrossRef CAS PubMed.
- Y. Tezuka, R. Kasimu, P. Basnet, T. Namba and S. Kadota, Chem. Pharm. Bull., 1997, 45, 1306 CrossRef CAS.
- B. Y. Lam, A. C. Lo, X. Sun, H. W. Luo, S. K. Chung and N. J. Sucher, Phytomedicine, 2003, 10, 286 CrossRef CAS PubMed.
- (a) Y. Yoon, Y. O. Kim, W. K. Jeon, H. J. Park and H. J. Sung, J. Ethnopharmacol., 1999, 68, 121 CrossRef CAS; (b) H. J. Sung, S. M. Choi, Y. Yoon and K. S. An, Exp. Mol. Med., 1999, 31, 174 CrossRef CAS PubMed; (c) M. A. Mosaddik, Phytomedicine, 2003, 10, 682 CrossRef CAS PubMed.
- M. Sairafianpour, J. Christensen, D. Stærk, B. A. Budnik, A. Kharazmi, K. Bagherzadeh and J. W. Jaroszewski, J. Nat. Prod., 2001, 64, 1398 CrossRef CAS PubMed.
- (a) Y. Gong, Y. Li, Y. Lu, L. Li, H. Abdolmaleky, G. L. Blackburn and J. R. Zhou, Int. J. Cancer, 2011, 129, 1042 CrossRef CAS PubMed; (b) D. S. Shin, H. N. Kim, K. D. Shin, Y. J. Yoon, S. J. Kim, D. C. Han and B. M. Kwon, Cancer Res., 2009, 69, 193 CrossRef CAS PubMed.
- D. S. Lee and S. D. Hong, J. Microbiol. Biotechnol., 1998, 8, 89 CAS.
- S. Y. Ryu, C. O. Lee and A. U. Choi, Planta Med., 1997, 63, 339 CrossRef CAS PubMed.
- J. H. Sun, M. Yang, X. C. Ma, J. Kang, J. Han and D. A. Guo, J. Asian Nat. Prod. Res., 2009, 11, 482 CrossRef PubMed.
- (a) P. J. Davis, Dev. Ind. Microbiol., 1988, 29, 197 CAS; (b) D. Zhang, Y. Yang, J. E. Leakey and C. E. Cerniglia, FEMS Microbiol. Lett., 1996, 138, 221 CrossRef CAS.
- (a) R. V. Smith and J. P. Rosazza, Arch. Biochem. Biophys., 1974, 161, 551 CrossRef CAS; (b) R. V. Smith and J. P. Rosazza, J. Pharm. Sci., 1975, 64, 1737 CrossRef CAS; (c) S. Asha and M. Vidyavathi, Biotechnol. Adv., 2009, 27, 16 CrossRef CAS PubMed.
- (a) S. Block, P. Gerkens, O. Peulen, O. Jolois, M.-P. Mingeot-Leclercq, M.-C. De Pauw-Gillet and J. Quetin-Leclercq, Anticancer Res., 2005, 25, 363 CAS; (b) L. A. Mistcher, G. S. R. Rao, T. Veysoglu, S. Drake and T. Haas, J. Nat. Prod., 1983, 46, 745 CrossRef; (c) E. L. Ghisalberti, Fitoterapia, 1997, 68, 303 CAS; (d) B. M. Fraga, Phytochem. Anal., 1994, 5, 49 CrossRef CAS.
- B. M. Fraga, V. González-Vallejo and R. Guillermo, J. Nat. Prod., 2011, 74, 1985 CrossRef CAS PubMed.
- A. Leverrier, M. T. Martin, C. Servy, J. Ouazzani, P. Retailleau, K. Awang, M. R. Mukhtar, F. Gueritte and M. Litaudon, J. Nat. Prod., 2010, 73, 1121 CrossRef CAS PubMed.
- E. A. Silva, J. A. Takahashi and A. B. Oliveira, J. Braz. Chem. Soc., 2002, 13, 101 CrossRef CAS PubMed.
- B. M. Fraga, M. G. Hernández, C. Fernández and J. M. Arteaga, Phytochemistry, 1987, 26, 775 CrossRef CAS.
- (a) S. Suebsasana, P. Pongnaratorn, J. Sattayasai, T. Arkaravichien, S. Tiamkao and C. Aromdee, Arch. Pharmacal Res., 2009, 32, 1191 CrossRef CAS PubMed; (b) N. L. Daló, M. C. Sosa-Sequera and A. Usubillaga, Invest. Clin., 2007, 48, 349 Search PubMed.
- B. C. Cavalcanti, L. V. Costa-Lotufo, M. O. Morales, R. R. Burbano, E. R. Silveira, K. M. Cunha, V. S. Rao, D. J. Moura, R. M. Rosa, J. A. Henriques and C. Pessoa, Food Chem. Toxicol., 2006, 44, 388 CrossRef CAS PubMed.
- B. M. Fraga, I. de Alfonso, V. González-Vallejo and R. Guillermo, Tetrahedron, 2010, 66, 227 CrossRef CAS PubMed.
- S. El-Sharkawy and Y. Abul-Hajj, J. Nat. Prod., 1987, 50, 520 CrossRef CAS.
- J. Zhan, H. Guo, L. Ning, Y. Zhang and D. Guo, Planta Med., 2006, 72, 346 CrossRef CAS PubMed.
- B. M. Fraga, L. Alvarez and S. Suárez, J. Nat. Prod., 2003, 66, 327 CrossRef CAS PubMed.
- B. M. Fraga, R. Guillermo and M. G. Hernández, J. Nat. Prod., 2004, 67, 64 CrossRef CAS PubMed.
- (a) M. Grande, M. Segura and B. Mancheño, J. Nat. Prod., 1986, 49, 259 CrossRef CAS; (b) M. Grande, M. J. Macías, B. Mancheño, M. Segura and A. Zarzo, J. Nat. Prod., 1991, 54, 866 CrossRef CAS.
- (a) D. T. Dennins, C. D. Upper and C. A. West, Plant Physiol., 1965, 40, 948 CrossRef PubMed; (b) B. E. Cross and P. L. Myers, Phytochemistry, 1969, 8, 79 CrossRef CAS.
- (a) B. M. Fraga, P. González, M. G. Hernández, F. G. Tellado and A. Perales, Phytochemistry, 1986, 25, 1235 CrossRef CAS; (b) B. M. Fraga, M. G. Hernández and P. González, Phytochemistry, 1992, 31, 3845 CrossRef CAS.
- F. Nagashima, M. Kondoh, T. Uematsu, A. Nishiyama, S. Saito, M. Sato and Y. Asakawa, Chem. Pharm. Bull., 2002, 50, 808 CrossRef CAS.
- (a) Z. J. Jia, J. G. Shi and Y. Li, J. Nat. Prod., 1994, 57, 811 CrossRef CAS; (b) Q. S. Zhao, Z. W. Lin, B. Jiang, J. Wan and H. D. Sun, Phytochemistry, 1999, 50, 123 CrossRef CAS.
- J. M. C. Geuns, Phytochemistry, 2003, 64, 913 CrossRef CAS.
- E. Koyama, K. Kitazawa, Y. Ohori, O. Izawa, K. Kakegawa, A. Fujino and M. Ui, Food Chem. Toxicol., 2003, 41, 359 CrossRef CAS.
- P. Pariwat, S. Homvisasevongsa, C. Muanprasat and V. Chatsudthipong, J. Pharmacol. Exp. Ther., 2008, 324, 798 CrossRef CAS PubMed , and the references cited therein.
- H. G. W. Leuenberger, Pure Appl. Chem., 1990, 62, 753 CrossRef CAS.
- F. L. Hsu, C. J. Hou, L. M. Yang, J. T. Cheng, P. C. Liu and S. J. Lin, J. Nat. Prod., 2002, 65, 273 CrossRef CAS PubMed.
- (a) J. R. Hanson, Nat. Prod. Rep., 1992, 9, 139 RSC; (b) B. H. de Oliveira and R. A. Strapasson, Phytochemistry, 1996, 43, 393 CrossRef CAS; (c) B. H. de Oliveira, M. C. dos Santos and P. C. Leal, Phytochemistry, 1999, 51, 737 CrossRef CAS.
- S. F. Chang, L. M. Yang, C. H. Lo, J. H. Liaw, L. H. Wang and S. J. Lin, J. Nat. Prod., 2008, 71, 87 CrossRef CAS PubMed.
- S. F. Chang, L. M. Yang, F. L. Hsu, J. Y. Hsu, J. H. Liaw and S. J. Lin, J. Nat. Prod., 2006, 69, 1450 CrossRef CAS PubMed.
- T. Akihisa, Y. Hamasaki, H. Tokuda, M. Ukiya, Y. Kimura and H. Nishino, J. Nat. Prod., 2004, 67, 407 CrossRef CAS PubMed.
- S. F. Chang, B. H. Chou, L. M. Yang, F. L. Hsu, W. K. Lin, Y. Ho and S. J. Lin, Bioorg. Med. Chem., 2009, 17, 6348 CrossRef CAS PubMed.
- O. I. Militsina, G. I. Kovyljaeva, G. A. Bakaleynik, I. Y. Strobykina, V. E. Kataev, V. A. Alfonsov, R. Z. Musin, D. V. Beskrovny and I. A. Litvinov, Mendeleev Commun., 2005, 15, 27 CrossRef PubMed.
- B. H. Chou, L. M. Yang, S. F. Chang, F. L. Hsu, C. H. Lo, J. H. Liaw, P. C. Liu and S. J. Lin, J. Nat. Prod., 2008, 71, 602 CrossRef CAS PubMed.
- B. H. Chou, L. M. Yang, S. F. Chang, F. L. Hsu, C. H. Lo, W. K. Lin, L. H. Wang, P. C. Liu and S. J. Lin, Phytochemistry, 2009, 70, 759 CrossRef CAS PubMed.
- C. D. Hufford, R. D. Guerrero and N. J. Doorenbos, J. Pharm. Sci., 1976, 65, 778 CrossRef CAS.
- A. S. Lamm, W. F. Reynolds and P. B. Reese, Phytochemistry, 2006, 67, 1088 CrossRef CAS PubMed.
- A. R. M. Chen, P. L. D. Ruddock, A. S. Lamm, W. F. Reynolds and P. B. Reese, Phytochemistry, 2005, 66, 1898 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |