
Dual functionality of conjugated polymer nanoparticles as an anticancer drug carrier and a fluorescent probe for cell imaging†
Abstract
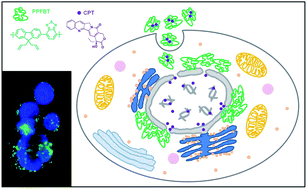
Multifunctional nanoparticles based on a green emitting, hydrophobic conjugated polymer, poly[(9,9-bis{propeny}fluorenyl-2,7-diyl)-co-(1,4-benzo-{2,1,3}-thiodiazole)] (PPFBT), that acts both as a fluorescent reporter and a matrix to accommodate an anti-cancer compound, camptothecin (CPT), were prepared, characterized and their potential as a fluorescent probe for cell imaging and as a drug delivery vehicle were evaluated via in vitro cell assays. The cell viability of human hepatocellular carcinoma cell line (Huh7) was investigated in the absence and presence of CPT with sulforhodamine B (SRB) and real-time cell electronic sensing (RT-CES) cytotoxicity assays.
 Please wait while we load your content...
Please wait while we load your content...

