
Carbon dioxide absorption by hydroxyalkyl amidines impregnated into mesoporous silica: the effect of pore morphology and absorbent loading†
Abstract
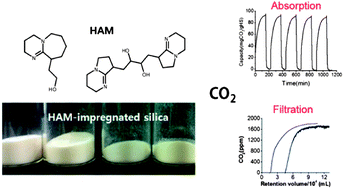
Hydroxyalkyl amidine (HAM) derivatives react with CO2 in a quantitative manner under dry conditions near room temperature, and then release it at a moderate temperature of approximately 60 °C. In the present study, HAM compounds were impregnated into porous silica particles to obtain solid CO2 sorbents. This process produced a large surface area for reaction between CO2 and the absorbents for enhanced CO2 capture efficiency. Several types of mesoporous silicas that differed in pore size, pore volume, and particle size and shape were investigated in order to identify the optimum processing parameters for preparation of hybrid materials with the best possible CO2 capture performance. The amount of HAMs loaded into the silica was optimized so as to retain the open porous structure for efficient transport of gas molecules, while providing a large surface area of reactive material. We have demonstrated that HAM-loaded silica could be used as an effective CO2 filter for dilute gas mixtures such as air.
 Please wait while we load your content...
Please wait while we load your content...

