
Stereoselective total synthesis of (+)-hyptolide†
Abstract
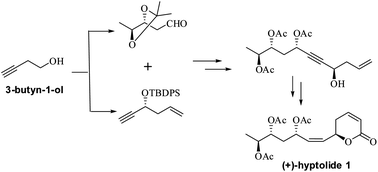
Stereoselective synthesis of the naturally occurring 6-membered lactone hyptolide 1 is described. The main feature of the synthesis is the utility of the hitherto unexplored alkyne fragment derived from commercially available 3-butyn-1-ol (homopropargylic alcohol).
 Please wait while we load your content...
Please wait while we load your content...

