
Characterization of the reaction products, kinetics and mechanism of oxidation of the drug captopril by platinum(iv) complexes†
Abstract
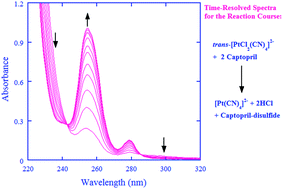
Captopril, the first pharmaceutical drug designed, synthesized, and used primarily for the treatment of hypertension and congestive heart failure, is an angiotensin-converting-enzyme inhibitor and is also an antioxidant. On the other hand, the kinetic and mechanistic aspects for the oxidation of captopril are not well understood. The oxidation of captopril by cisplatin prodrug and a model compound, cis-[Pt(NH3)2Cl4] and trans-[PtCl2(CN)4]2−, was thus investigated in this work. A stopped-flow spectrometer was employed to follow the oxidation kinetics over a wide pH-range under the pseudo first-order conditions of [captopril] ≫ [Pt(IV)]. The oxidation by the Pt(IV) complexes displayed a second-order character, first-order each in [Pt(IV)] and in [captopril], whereas the metal-ion-catalyzed autooxidation made a very minor contribution to the overall kinetics in acidic media and was negligible in neutral media. Captopril was oxidized to form the captopril-disulfide as identified by ESI mass spectrometry under the conditions of the kinetic measurements. In the proposed reaction mechanism, the Pt(IV) complexes are reduced by the three protolytic captopril species in parallel as rate-determining steps, generating reactive species of chlorothiol and/or sulfenylchloride. The reactive species will be rapidly trapped, either directly or indirectly, by another molecule of captopril to form captopril-disulfide. The rate constants for the rate-determining steps have been derived, demonstrating that the fully deprotonated captopril is about 105 to 106 times more reactive than its corresponding thiol form toward the Pt(IV) complexes.
 Please wait while we load your content...
Please wait while we load your content...

