
Versatile metal complexes of 2,5-bis{N-(2,6-di isopropylphenyl)iminomethyl}pyrrole for epoxide–CO2 coupling and ring opening polymerization of ε-caprolactone†
Abstract
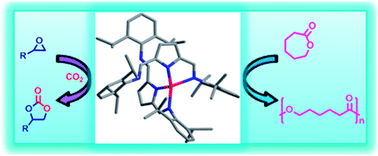
The catalytic activities of metal complexes of 2,5-bis{N-(2,6-diisopropylphenyl)iminomethyl}pyrrole 1 for epoxide–CO2 coupling and ring opening polymerization (ROP) of ε-caprolactone (CL) were explored. The Zn(II) 2, Cd(II) 3,4 and Cu(II) 5 complexes of 1, in the presence of tetrabutylammonium bromide (TBAB) as co-catalyst, effectively catalyzed the epoxide–CO2 coupling reaction at 1 atm of CO2 to produce cyclic carbonates. In addition, the catalytic activities of these complexes (2–5) were investigated for the ROP of CL in the presence and absence of benzyl alcohol (BnOH). All these complexes were active for ROP of CL at mild temperature. In particular, the complexes 2 and 5 exhibited remarkable catalytic activities for ROP of CL at 25 °C in toluene in the presence of BnOH. The ROP of CL catalyzed by 2 and 5 complexes, not only proceeded through living polymerization but also immortal.
 Please wait while we load your content...
Please wait while we load your content...

