
Photoinduced solid state keto–enol tautomerization of 2-(2-(3-nitrophenyl)-4,5-diphenyl-1H-imidazol-1-yloxy)-1-phenylethanone†
Abstract
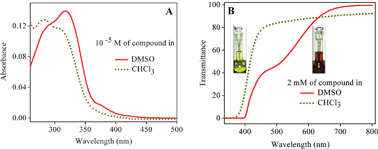
Excited state intramolecular proton transfer (ESIPT) plays an important role in biological systems and has also recently found applications in electronic devices such as transducers, switches etc. In this paper we report the synthesis and solid state photochromic behavior of 2-(2-(3-nitrophenyl)-4,5-diphenyl-1H-imidazol-1-yloxy)-1-phenylethanone (II) due to ESIPT. Compound II exhibits yellow color in dark and red color in light, with the yellow form attributed to the keto derivative and the red form assigned to its enol derivative The color change in the presence of light is thus attributed to the keto–enol tautomerism through ESIPT. The color change from yellow to red is a photochemical process which thermally decays to the yellow form in the dark. The solid state stability of the enol form upon phototautomerization of the keto form is a noteworthy phenomenon, and its stability has been substantiated by our experimental findings. In the solution state, the yellow form (keto) is stable in chloroform while the red form (enol) is stable in DMSO. Theoretical calculations have been performed to understand the geometries and electronic transitions of the keto and enol forms. In addition, ground and excited state equilibrium constants for the keto–enol tautomerism were calculated.
 Please wait while we load your content...
Please wait while we load your content...

