
Influence of Al doping on microstructural, optical and photocatalytic properties of sol–gel based nanostructured zinc oxide films on glass†
Abstract
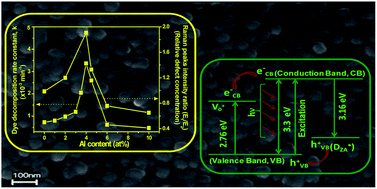
Al doped nanostructured zinc oxide thin films (thickness, 165 ± 5 nm) on silica glass were prepared from zinc acetate based solutions of varying dopant content (0 to 10 at% with respect to Zn). X-ray diffraction confirmed the presence of nanocrystalline hexagonal ZnO. On increasing the doping level, we observed a change in ZnO morphology (spherical, hexagon and plate-shaped) under field emission scanning and transmission electron microscopes and a gradual decrease of ZnO crystallite size (14.0 to ∼10 nm) vis-à-vis an enhancement of direct band gap energy of the films. Root mean square film surface roughness and chemical state of elements were studied by atomic force microscopy and X-ray photoelectron spectroscopy respectively. In addition to common intrinsic defects in ZnO, a defect (designated as D˙ZA, appearing as a paramagnetic singly negatively charged oxygen vacancy) was identified up to 4% doping from the appearance of photoluminescence emission at 398 nm and measurement of paramagnetic property of the films. Film photocatalytic activity towards Rhodamine 6G dye decomposition was performed under UV (254 nm) irradiation and the film with 4% doping (A4ZO) showed the highest value of first order decomposition rate constant. On increasing Al content, the trend of change of defect concentration (oxygen vacancies) analyzed by Raman spectra was found to be identical with the dye photodecomposition activity (PA) of the films. A major role of DZA˙ towards the PA was explored under visible light. We proposed the reaction mechanism of PA based on the experimental results. The A4ZO would be expected to decompose micro-organisms under visible light.
 Please wait while we load your content...
Please wait while we load your content...

