Coinage metals (Cu, Ag and Au) in the synthesis of natural products
K. C. Majumdar
* and
Biswajit Sinha
Department of Chemistry, University of Kalyani, Kalyani 741235, West Bengal, India. E-mail: kcm_ku@yahoo.co.in; kcm@klyuniv.ac.in
First published on 21st November 2013
Abstract
The chemistry of natural products has emerged as an extremely important area of contemporary research owing to their profound biological activities, complex molecular architecture with challenging stereochemistry and potential for use as medicine. Searches for efficient synthetic procedures for natural products, drugs, and materials with economical and ecological advantages are important areas of current research interest. This article describes the isolation, bioactivity and synthesis of several natural products and drugs by employing coinage metal-catalyzed/mediated reaction/strategy either in a sequential or a domino fashion. We have made an attempt to summarize the progress made in the area from 2008 onwards.
 K. C. Majumdar | Krishna C. Majumdar received Ph. D. from the University of Idaho. He had postdoctoral training in USA and Canada. He has been with the University of Kalyani since 1977 and is there presently as UGC Emeritus Fellow. He had also served North-Eastern Hill University as visiting Professor (1996), Indian Institute of Technology (Kharagpur) as Associate Professor on lien (1990–1991) and Tezpur University (2011) as Professor of Eminence. His research interests center around synthetic organic chemistry, design and synthesis of liquid crystals with over 400 publications. He received Chemical Research Society of India Medal (2004) and Indian Chemical Society Award (2006). |
 Biswajit Sinha | Biswajit Sinha received his B.Sc. in 2004 and M.Sc. in 2006 from the University of Kalyani. He then joined the research group of Prof. K. C. Majumdar at the University of Kalyani with a CSIR-NET fellowship. He was awarded his Ph. D. in 2013. He is mainly working on metal-mediated synthesis of heterocycles and molecular iodine-mediated synthesis of potentially bioactive heterocycles. |
1. Introduction
The chemistry of natural products has emerged as a broad and remarkable field of research due to their profound biological activities, complex molecular architecture with challenging stereochemistry and potential for medicinal use.1 Some of the natural products without known biological activity can readily be converted into biologically useful compounds of greater value by simple or multistep chemical conversions. So natural product synthesis is a highly challenging field in constant demand for improved protocols. The development of new approaches for their synthesis employing efficient and mild reaction conditions is currently a key area of research in natural products. The reaction protocols are often chosen in such a way that it can allow straightforward, scalable, and high-yield conversions with functional groups tolerance. Additionally, the reaction is designed to be applicable to complex substrates for easy introduction of certain functional groups. The complex structures and functionalities of these difficult targets require robust and selective methods for utilization. Several literature reviews have covered the total synthesis of natural products.2 Recently, catalysis by coinage metals which include copper (Cu), silver (Ag) and gold (Au) has emerged as a controlling tool for various C–C and C–X bond forming reactions.3 These reactions often proceed with interesting mechanistic pathways. Sometimes, coinage metals are used as cocatalysts i.e.; with the combination of other metals such as Pd–Cu, Pd–Ag, Rh–Ag, Pt–Ag, Au–Ag, and Mn–Cu. A number of literature reviews covered the examples where Cu, Ag, and Au metals are used as catalysts at key steps to generate bioactive natural products.4 The purpose of this review is to discuss the use of coinage metals as a useful catalyst for the synthesis of complex natural products. We have tried to include many recent examples from 2008 to till date but excluded the examples where the coinage metals were used as co-catalysts. We have also tried to discuss the mechanism of the reactions in brief wherever possible, to present an idea about the activation of the substrates by coinage metals and also probable reaction pathways.2. Copper-catalyzed synthesis of natural products
C–N, C–O, and C–C bond formation reactions made copper catalysis an emerging area of research. At present, copper-based reagents are frequently used as key steps in the total synthesis of a wide variety of natural products. The copper catalysts are highly tolerant to different functional groups and this has made them useful in the development of orthogonal catalytic systems for selective C–N or C–O bond formation.5 Copper catalysts are less costly and require mild reaction conditions. Recent advances in enantioselective copper-catalyzed reactions and C–H activations helped to meet the challenge of the synthesis of complex organic molecules.62.1. Cuprate reactions
(+)-Cassaine 4 is a nonsteroidal inhibitor of Na+, K+-ATPase. It possesses a pharmacological action similar to digitoxins, though their structures are quite different.7 It was isolated in 1935 from the bark of Erythrophleum guineense. The Turner group8 established its structure and also reported the only total synthesis of (+)-cassaine, in spite of their initial difficulty with the functionalization of rings B and C. (+)-Cassaine has been the subject of investigation in chemical biology because of its efficient cardiotonic properties, equal to that of digitalis glycosides.The TAC shape (trans-anti-cis) of enal 2 indicated that essentially only the R-face is sterically available for the 1,4-addition of a methyl group, leading to the desired R stereochemistry at C14. After several experimentats on models the addition was accomplished using copper cyanide, methyllithium and TMSCl at −30 °C and the desired enol ether 3a was obtained. The intermediate 3a is quite unstable and in order to avoid decomposition the enol ether was immediately converted to the corresponding stable triflic enol ether 3b with 54% yield using methyllithium and N-phenyltrifluoromethane sulfonimide in THF (Scheme 1).9
 | ||
| Scheme 1 | ||
The lipid phthiocerol dimycocerosate A (PDIM A) 5 is a wax which contains two tetramethyl substituted saturated acids, namely mycocerosic acid and phthiocerol.10 Minnaard et al. accomplished11 the first asymmetric synthesis of phthiocerol in 15 steps and 5.6% overall yield by applying the copper-catalyzed asymmetric conjugate addition reaction as a key step (Fig. 1).
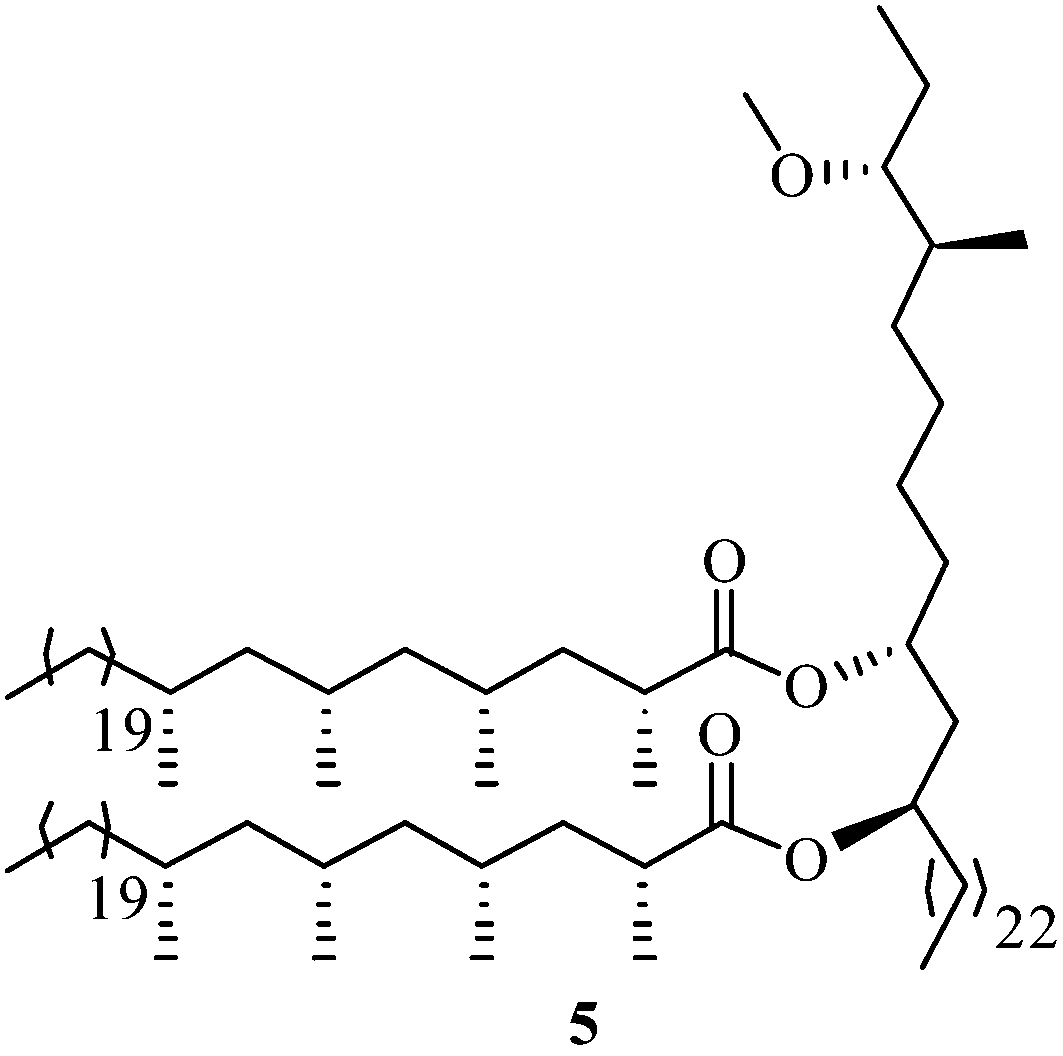 | ||
| Fig. 1 Lipid phthiocerol dimycocerosate A, PDIM A. | ||
Clavirolide C 6 is a member of the dolabellane family of diterpenes and has been isolated from the Pacific soft coral Clavularia viridis. The structure contains a characteristic trans-bicyclo[9.3.0]tetradecane core.12 The Hoveyda group reported a concise total synthesis of clavirolide C, demonstrating the utility of Cu-catalyzed ACA reactions (asymmetric conjugate addition) of unsaturated heterocycles with dialkyl zinc reagent in one of the steps (Fig. 2).13
 | ||
| Fig. 2 Dolabellane family diterpene, clavirolide C | ||
Sultriecin 10,14 an antitumor antibiotic as well as protein phosphatase 2A (PP2A) inhibitor, was isolated from the Streptomyces roseiscleroticus no. L827-7. Considering its functional biological activity and structural similarity to other natural products such as fostriecin and cytostatin the Boger group suggested that 10 would also be a selective PP2A inhibitor. So they developed the first total synthesis of 10, established its stereochemistry and also established that it represents an effective inhibitor of PP2A. Incorporation of the sensitive Z,Z,E-triene tail was found to be crucial. This was successfully accomplished in 85% yield (>5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) by the copper-catalyzed reaction (CuI-PBu3, Et2O, −78 °C, 15 min) of the compound 7 followed by slow addition of the aldehyde 8 (Et2O, −78 °C, 1 h) (Scheme 2).15
:1 dr) by the copper-catalyzed reaction (CuI-PBu3, Et2O, −78 °C, 15 min) of the compound 7 followed by slow addition of the aldehyde 8 (Et2O, −78 °C, 1 h) (Scheme 2).15
 | ||
| Scheme 2 | ||
Spring et al. disclosed16 a concise de novo total synthesis of deoxyschizandrin based on a double organocuprate oxidation strategy. Moreover, they also performed biological tests to explore the ability of deoxyschizandrin and synthetic precursors for use as an inhibitor of the proliferation of a human cancer cell line.17 The natural product deoxyschizandrin 15 is a potential therapeutic agent against cancer. It is used as a traditional Chinese medicine.18 Deoxyschizandrin was isolated from the seed oil of Schisandra chinensis.19 It shows biological activities including antiviral20 and anti-inflammatory activities.21
Attempts at homo-coupling of the iodo-derivative 11 using the lithio-cuprate via metalation with tBuLi were unsuccessful.22 Milder metalation conditions proved useful, at a low temperature. Functional group-tolerant I/Mg exchange23 on 11 produced an alkenyl Grignard reagent. This Grignard reagent was transmetalated to the magnesio-cuprate and oxidized to furnish 13 in good yield. This successful result with a highly sterically hindered substrate demonstrates the utility of alkenyl cuprate oxidation under difficult conditions. Moreover, only one geometrical isomer was produced. The compound 13 on reduction with H2, Rh/C followed by halogenation afforded the next coupling precursors 14. With these intermediates (X = Br, I) in hand the key organocuprate oxidative intramolecular biaryl bond-forming reaction was attempted. Optimal results were achieved using iodo derivative (R,S)-14b. The intermediate 14b was treated with isopropyl-magnesium chloride, followed by CuBr·SMe2 (transmetalation) and oxidant 12 (intramolecular cuprate oxidation) to afford the natural product (−)-deoxyschizandrin. The reaction proceeded in good yield avoiding both the high dilution reaction condition and formation of a dimer side product (Scheme 3).
 | ||
| Scheme 3 | ||
2.2. Cu-catalyzed C–O bond formation
Azithromycin 20, a 15-membered macrolide, is a semi-synthetic antibiotic. This is derived from erythromycin A by a sequence of reactions, viz., oximation, Beckmann rearrangement, reduction, and N-methylation.24 Azithromycin, the first azalide, shows the highest antibacterial activity among its family members.Using the Cu(OTf)2-CuO-catalyzed coupling protocol the branched neutral sugar was attached to 17 using 8 equivalents of 2-thiopyridyl cladinoside 18.25 A 6:1 separable anomeric mixture of the TBS-protected azalide was obtained in favor of the desired β-anomer 19 in 53% yield along with 24% of the starting lactone 17. Removal of the α-anomer afforded the purified β-anomer which was finally desilylated to give azithromycin (20) in 89% yield. Thus a highly stereoselective total synthesis of azithromycin 20 was achieved in a linear sequence of 18 steps from the readily available chiral building block 16 (Scheme 4).26
 | ||
| Scheme 4 | ||
A copper-mediated Ullmann-type etherification reaction was carried out as the key step for the total synthesis of Hirsutellone B 23. It was isolated from the insect pathogenic fungus Hirsutella nivea BCC 2594. It is found to exhibit antituberculosis activity against Mycobacterium tuberculosis H37Ra with a minimum inhibitory concentration (MIC) value of 0.78 µg mL−1.27 The main structural core of Hirsutellone B consists of the highly strained 13-membered macrocycle including a bent benzene ring. The construction of this molecular framework is found to be too difficult. Only one asymmetric total synthesis has been reported to date.28 Thus the structural complexity as well as its intriguing biological activity attracted much attention from synthetic chemists and pharmaceutical researchers.
Uchiro et al. used29 the intramolecular Ullmann-type reaction of the compound 21 to construct the highly strained 13-membered macrocyclic intermediate 22. The desired cyclization product was obtained in 42% yield by increasing the reaction temperature to 160 °C. The standard Buchwald conditions30 were found to be ineffective for this reaction. Moreover, the strategy provided the first successful example of a macrocyclization between an aliphatic alcohol and an aryl halide (Scheme 5).
 | ||
| Scheme 5 | ||
The Takahashi group developed31 a short but efficient method for the synthesis of vialinin B 24 using a Cu-mediated Ullmann reaction in one of the steps (Fig. 3). Vialinin B is a highly oxygenated dibenzofuran possessing p-hydroxyphenyl and phenylacetoxy groups. It shows potent inhibitory activity against tumor necrosis factor (TNF)-α production in rat basophilic leukemia (RBL-2H3) cells (IC50 = 20 pM vs. 0.25 nM for FK-506).32
 | ||
| Fig. 3 Highly oxygenated dibenzofuran vialinin B | ||
An efficient copper-catalyzed Ullmann coupling reaction was used in one of the steps for the synthesis of eleven-membered biaryl ether lactones of the aspercyclide family 25a–c (Fig. 4).33 These metabolites interfere with the binding of immunoglobulin E antibodies (IgEs) to the human high-affinity IgE receptor FceRI located in the membrane of mast cells and basofils and are potent in the treatment of allergic disorders including asthma.34
 | ||
| Fig. 4 Eleven-membered biaryl ether lactones of the aspercyclide family | ||
2.3. Cu-catalyzed C–N coupling reactions
Mycestericins, potent immunosuppressants, were isolated from the culture broth of Mycelia sterilia.35 The mode of action of mycestericins was found to be closely related to that of myriocin.36 The Shibasaki group developed an amide ligand-based unique ternary catalytic system using La(NO3)3·6H2O and D-valine tert-butyl ester H-D-Val-OtBu. This was specifically efficient for the catalytic asymmetric amination of highly coordinative substrates exhibiting multiple coordination modes. Later on they realized that the ternary catalytic system could be highly useful for a concise enantioselective total synthesis of mycestericins F 30 and G 31.37 Thorough experimentation also indicated that changing the metal catalyst from lanthanum to copper along with the ligand 28 was much better and afforded the desired product 29 in quantitative yield with 95% ee at room temperature (Scheme 6).38 | ||
| Scheme 6 | ||
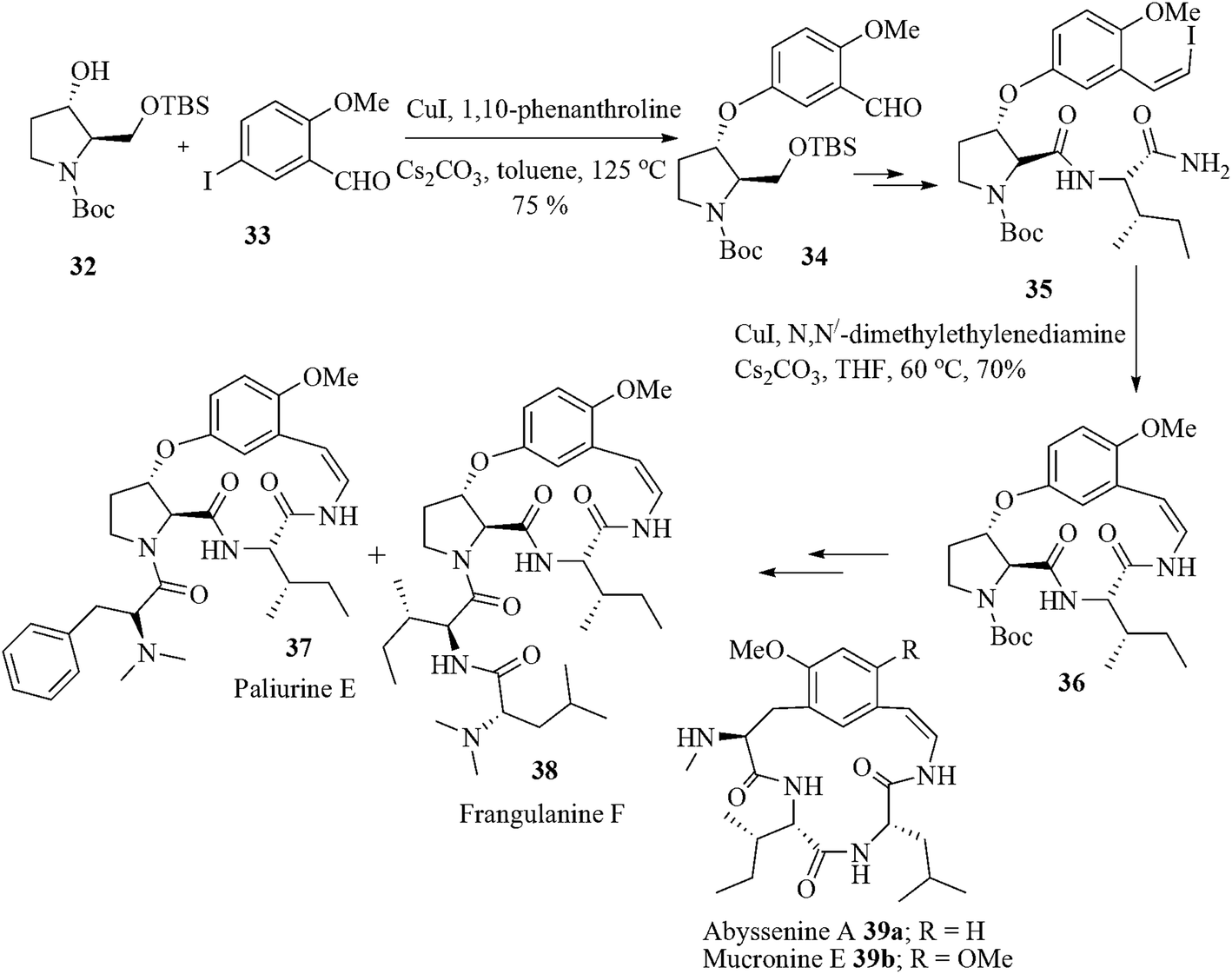
Cyclopeptide alkaloids are cyclic polyamide bases of plant origin and are found in the roots, leaves, and bark of a number of plants. Among them paliurine E 37, frangulanine F 38 and abyssenine A 39a possess 15- or 13-membered cyclic rings, respectively, along with an aromatic ring. The rest of the macrocycle is made up of a peptide unit that is connected to the aromatic ring in a 1,3-orientation via enamide and alkyl aryl ether linkages.39 They are used as remedies for diarrhea, dysentery or insomnia.40 Moreover, they also display sedative/stimulant, antibacterial, antifungal and antiplasmodial properties.41 The impressive biological properties along with interesting structures attracted the attention of synthetic organic chemists resulting in studies directed towards the synthesis of this class of compounds.42 All the protocols suffered from some synthetic challenges including elaboration of the aryl alkyl ether, formation of the strained macrocycle, and introduction of the enamide unit. The protocol developed by Joullie et al., Han et al. and Zhu et al. enriched the total synthesis chemistry of these cyclopeptide alkaloids but the elaboration of the enamide moiety by different stepwise elimination methods after macrocyclization decreased the overall synthetic efficiency.43 In this respect the work of Evano et al.44 is noteworthy. The group designed their strategy in such a way that the common key intermediate 36 could individually be prepared through Bronsted acid-catalyzed cycloaddition, Pd-catalyzed intramolecular oxidative amidation, ring closing metathesis or copper-mediated intramolecular amidation reaction from their corresponding acyclic starting materials. After careful experimentation they observed that copper-mediated intramolecular amidation reaction is superior compared to other methods and afforded a good yield of the cyclic intermediate 36. This alternative cyclization strategy is based on a copper-catalyzed intramolecular amidation of a vinyl iodide in a macrocyclization step. This has been successfully utilized for an efficient asymmetric synthesis of paliurine F, abyssenine A and mucronine E.
The compound 32 prepared from boc-protected D-serine was treated with 33 under the optimized conditions using copper(I) iodide and 1,10-phenanthroline in the presence of cesium carbonate in toluene at 125 °C. The intermediate compound 34, in few steps, afforded the macrocycle precursor 35. The iodo amide 35 afforded the 13-membered ring 36 in 70% yield on treatment with catalytic copper(I) iodide and N,N-dimethylethylenediamine in THF under high-dilution conditions at 60 °C. The compound 36, in few steps, afforded the alkaloids paliurine E 37 and frangulanine F 38. The interesting feature of this approach is the mild intramolecular amidation protocol which proceeds without any epimerization at the two amino acid stereocenters or isomerization of the (Z)-vinyl iodide. The reaction also avoids the dimerization or formation of higher oligomers. This macrocycle forming protocol is highly useful for the synthesis of other subclasses of cyclopeptide alkaloids such as abyssenine A (39a) and mucronine E (39b) using appropriate starting materials (Scheme 7).
 | ||
| Scheme 7 | ||
A copper(II)-catalyzed enantioselective intramolecular alkene carboamination reaction was employed for the enantioselective total synthesis of (S)-(+)-tylophorine 40 in eight steps from commercially available 3,4-dimethoxybenzyl alcohol. The phenanthroindolizidine alkaloid (R)-tylophorine was isolated from Tylophora indica, which is a potent cancer cell growth inhibitor. This also shows anti-inflammatory, anti-ameobicidal and anti-viral activities.45 (S)-Tylophorine is the unnatural enantiomer but it is a more potent inhibitor of cancer cell growth than (R)-tylophorine. Retrosynthetically, the Chemler group envisaged that (+)-tylophorine could be synthesized from the sultam 41 which could be prepared by the copper(II)-catalyzed tandem enantioselective carboamination reaction of compound 42. Thus they utilized the copper(II) complexes in efficient intramolecular carboamination of terminal alkenes46 to access the polycyclic aromatic nitrogen heterocycles. This methodology was applied to the sulfonamide 42 which underwent enantioselective carboamination to provide sultam 41. The sultam 41, in few steps, afforded the tylophorine 40. The sulfonamide serves a dual purpose in this reaction. It enhanced the reactivity compared to the corresponding amide and was also used as a handle for enantioinduction (Scheme 8).47
 | ||
| Scheme 8 | ||
The reaction is supposed to proceed via initial nitrogen–copper(II) bond formation (compound 44) followed by intramolecular migratory insertion and addition to the aromatic ring by a radical process to produce radical 46. The ortho-addition preference indicates that a radical character may be present in the aromatic addition step. An alternative reaction sequence was also proposed by the research group which may involve one-electron oxidation of the nitrogen to produce compound 47 followed by 5-exo-trig intramolecular ring closure to generate 48. Subsequent addition of the primary carbon-based radical onto the aromatic ring, followed by loss of a hydrogen radical, may provide 41 (Scheme 9).
 | ||
| Scheme 9 | ||
The uridylpeptide antibiotic pacidamycin D 53 was isolated from the fermentation broth of the Streptomyces coeruleorubiduns strain and showed potent and selective antibacterial activity against strains of Pseudomonas (MIC 1.5–12.5 µg mL−1).48 The phospho-MurNAc-pentapeptide transferase (MraY),49 the biological target of the pacidamycins is responsible for the formation of lipid I in the peptidoglycan biosynthetic pathway.50 The MraY has been considered as a potential target for the development of general antibacterial agents as it is an essential enzyme in bacteria.51 Therefore, uridylpeptide antibiotics have been considered to be possible antibacterial agents effective against P. aeruginosa. The main architecture of the compound 53 possesses a Z-oxyenamide moiety, which is chemically labile and perhaps for this reason no total synthesis of 53 has yet been accomplished. Therefore, it is a challenging chemical structure to construct.
The Matsuda group undertook the challenge to accomplish the total synthesis. From the retrosynthetic analysis they realized that the key step is an efficient and stereocontrolled construction of the intermediate 52 using a copper-catalyzed cross coupling52 of the Z-oxyvinyl iodide 50 with the highly functionalized tetrapeptide carboxamide 49. The key coupling of 50 with the tetrapeptide carboxamide 49 was investigated. Initially, the iodide 50 was reacted with 49 under the following conditions: 0.2 equiv. of CuI, 0.4 equiv. of MeNHCH2CH2NHMe, Cs2CO3, THF, 70 °C. Unfortunately, most of the iodide remained unreacted affording only a trace amount of the desired coupling product 52. The tetrapeptide 49 was consumed, and a cyclic side product was obtained from the reaction mixture. Generally, the copper-mediated C–N cross-coupling reaction proceeds via initial formation of the nitrogen–copper complex and an oxidative insertion into the halide followed by reductive elimination.53 It was suggested that if the oxidative insertion is slow, the nitrogen atom, activated by the formation of the carboxamide–copper(I) complex, may react with the tBu ester at the C-terminus to form the cyclic product. So an alternative strategy was designed and the approach of the nitrogen atom to the tBu ester was suppressed by increasing the size of the ligand by using ligands such as 51 coordinating to the copper atom. As expected, the use of the ligand 51 resulted in an increased yield. Thus the iodide 50 and the tetrapeptide 49 were coupled using 0.8 equiv. of CuI, 1.6 equiv. of ligand 51, Cs2CO3 in THF at 70 °C to afford the fully protected pacidamycin D 52 in 82% yield which after deprotection afforded the pacidamycin D 53 in 30% yield (Scheme 10).54
 | ||
| Scheme 10 | ||
Gerwick and coworkers isolated palmyrolide A 56a, a neuroactive macrolide from a cyanobacterial assemblage comprised of Leptolyngbya and Oscillatoria species.55 Biologically, it is a potent inhibitor of calcium ion oscillations in murine cerebrocortical neurons and possesses sodium ion channel blocking ability in neuroblastoma cells. The lactone was resistant to hydrolysis under a variety of reaction conditions. Attempts to degrade the macrolide into acyclic fragments were unsuccessful. Thus the determination of absolute stereochemistry could not be accomplished. The Murata J-based configurational analysis56 on the macrocycle itself was performed to determine the relative stereochemistry between the C(5) methyl and the C(7) tBu centers that, in combination with NOE correlations, indicated the relationship between C(5) and C(7) to be syn. Recently, Maio et al. synthesized57 the 15-membered ring of palmyrolide A, the trans-N-H enamide macrocycles in modest yield using high-dilution modification to the copper(I) iodide/cesium carbonate condition developed by Buchwald.58 A comparison of the reported spectra of palmyrolide A was made with that of the compound (+)-56a. Though compound (+)-56a was found to possess the natural C(10)-R stereochemistry, the spectral data did not match the literature values reported by Gerwick. On the other hand, a complete match of 1H and 13C NMR data of palmyrolide A was observed with that of the macrolide (+)-56b, where the C(10) methyl group is inverted relative to the natural macrolide. Finally, the optical rotation comparison confirmed that the synthesized enantiomer is enantiomer of (−)-palmyrolide A, (+)-ent-palmyrolide A (+)-56b {[α]D = +23 (c = 0.65, CHCl3), lit. [α]D = −29 (c = 0.9, CHCl3)}. Importantly, it established that the stereochemical relationship between the C(5) methyl and the C(7) tert-butyl is anti, and not syn, that was originally proposed in the isolation report. Notably, the absolute stereochemistry at C(5) and C(7) found in (−)-palmyrolide A is opposite to that is found in apratoxin A (Scheme 11).59
 | ||
| Scheme 11 | ||
An efficient, Cu-catalyzed asymmetric synthesis of the cytotoxic natural product chaetominine 60 was achieved in 14 steps and 10% overall yield starting from D-tryptophan and L-alanine.60 The ABC-tricyclic ring system of the compound 60 can be easily installed by the use of a copper(I)-mediated cyclization of a sensitive, highly substituted iodotryptophenylalanine derivative. The tricyclic system 59 was further elaborated by diastereoselective oxidation and reduction reaction. This is the first example of an oxidative rearrangement yielding homochiral spirocyclic pyrrolidinyloxindoles. (−)-Chaetominine 60 was isolated from the solid-substrate culture of Chaetomium sp. IFB-E015, an endophytic fungus on apparently healthy Adenophora axilliflora leaves.61 This tripeptide consists of a strained tetracyclic framework which is only encountered in seven other metabolites, the kapakahines.62 Chaetominine is a potential agent against human leukemia K562 (21 nM) and colon cancer SW1116 (28 nM) cell lines with IC50 values smaller than that of 5-fluorouracil, one of the most frequently prescribed anticancer drugs.
The cyclization of iodotryptophanylalanine derivative 57 to form the six-membered ring 59 is found to be challenging. This involves the formation of an intermediate seven-membered ring metallacycle. Considerable experimentation showed that this reaction can be carried out by a combination of copper iodide, trans-N,N′-dimethylcyclohexane-1,2-diamine 58 and potassium phosphate at 110 °C in toluene to give the best results.63 Application of this condition furnished the cyclized intermediate 59 in 64% isolated yield, without epimerization along with 30% of the reduced by-product (deiodinated product) that could be resubmitted to the iodination–cyclization sequence. The formation of a six-membered ring from the acyclic secondary amide 57 required the use of a more efficient ligand. The reduced by-products could not be completely avoided but were minimized. Other metals such as palladium(0) or iron(III) were found to be completely inefficient promoters for this cyclization (Scheme 12).64
 | ||
| Scheme 12 | ||
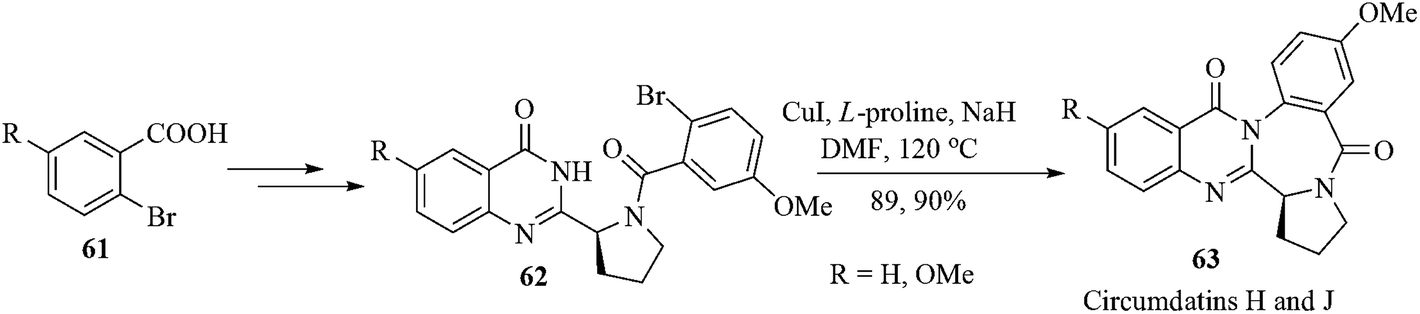
Circumdatins are the marine natural products from the fungi of the genus Aspergillus. Circumdatins possess antitumor, antifungal, insecticidal, and antibiotic activities.65 Among these (−)-circumdatins H and J were isolated from Aspergillus ochraceus and Aspergillus ostianus, respectively. The (−)-circumdatin H is an inhibitor of the mammalian mitochondrial respiratory chain, with an IC50 value of 1.5 µM.66 Though synthesis of (−)-circumdatins H and J have been achieved by using an intramolecular aza-Wittig reaction and reductive cyclization as the key steps,67 metal-catalyzed intramolecular N-arylation of quinazolinones was not studied.68 Recently, Argade et al. have succeeded in accomplishing an efficient convergent total synthesis of (−)-circumdatins H and J using copper-catalyzed intramolecular N-arylation of a quinazolinone nucleus that furnished the central benzodiazepine core unit of these natural products.69 The copper-catalyzed intramolecular N-arylation of the compounds 62a,b using L-proline as the ligand and sodium hydride as the base in DMF at 120 °C furnished the desired synthetic quinazolinones (−)-63a,b in excellent yields. The analytical and spectral data for the natural products (−)-circumdatins H and J (63a,b) completely matched with the reported data (Scheme 13).70
 | ||
| Scheme 13 | ||
2.4. Cu-catalysts as Lewis acid
Oximidine(II) (71) of the benzolactone enamide family of natural products, displays cytotoxicity at the ng mL−1 level in mutant rat fibroblasts.71 It is a selective inhibitor of mammalian vacuolar-type H + -ATPases (V-ATPases).72 Its promising anticancer activity attracted the attention of George et al. and encouraged them to try to develop a feasible synthetic route. Earlier the Porco73 and Molander74 groups in their total synthesis found that the major challenge in the synthesis of oximidine II is the formation of its strained 12-membered macrolactone core containing nine contiguous sp2-hybridized carbon atoms.75The copper catalyst was used for the construction of the central 12-membered macrolactone ring. The intermediate 66 was subjected to the reaction condition reported by Stryker et al. with the generation of [CuH(PPh3)]6 (ref. 76) to give only the reduced triene in 31% yield. Sodium formate was found to be the optimal source of hydride for this one-pot macrocyclization/reduction transformation and the triene macrocycle 70 was obtained in 67% yield. Reacting with Cu(OAc)2·H2O instead of sodium formate lowered yield (55%) of the desired triene 70 but affording a cleaner reaction. A mechanism based on the deuterium-labeling studies has been suggested for this reductive macrocyclization. The stereoselective ring closure of 66 was carried out by a Castro–Stephens ring closure reaction77 to produce the alkyne containing 8E macrocycle 67. The copper atom remained coordinated to the alkyne, and stabilized the C8–C9 E-olefin geometry and prevented the double-bond isomerization. Expulsion of CO2 from the formate generated the requisite Cu–H species 68. The species 68 underwent 1,2-addition across the triple bond to afford the vinyl copper intermediate 69 in which the copper is preferentially bound to C10. A C11–Cu intermediate must have formed as well since C11-deuterated products were also obtained. Finally, protonation of the copper metal species yielded the reduced macrocycle 70. This total synthesis provided oximidine II in a total of 14 steps and in 9.2% overall yield (Scheme 14).
 | ||
| Scheme 14 | ||
The copper-catalyzed stereoselective vinyl oxirane ring expansion reaction was applied as a key step for the formation of the natural product anti cancer agent varitriol 72 (Fig. 5).78
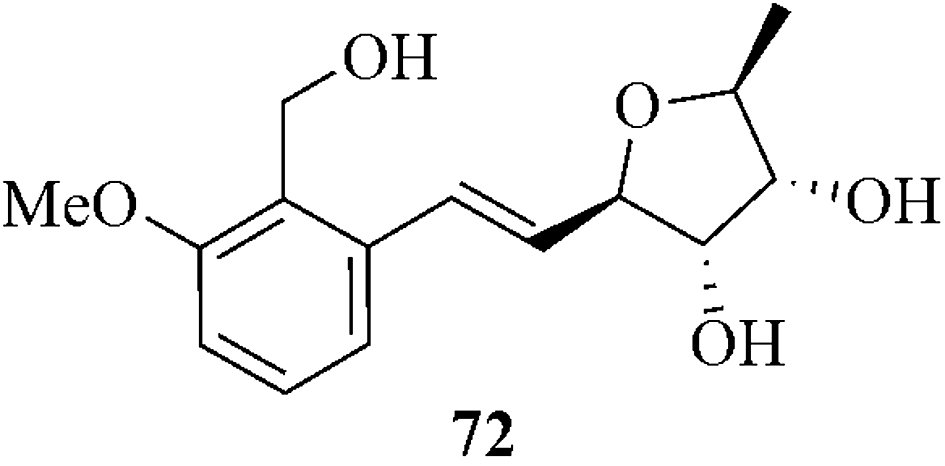 | ||
| Fig. 5 Anti cancer agent (+)-varitriol | ||
The same group also reported a new copper-catalyzed ring expansion of vinyl oxiranes using commercially available, air-stable copper(II) catalysts and applied the same to access natural products including the tetrahydrofuran-based anticancer agent (+)-goniothalesdiol 77.79 Highly enantiopure cis-2,5-dihydrofuran 76 was obtained from the chiral oxirane 74 in 70% yield. (Scheme 15).80
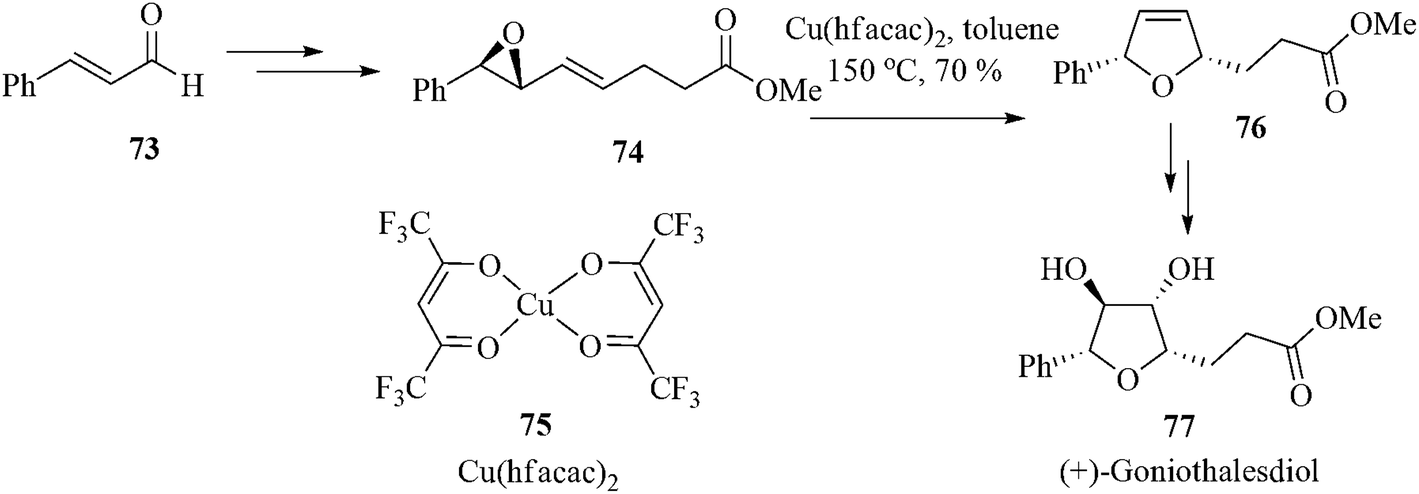 | ||
| Scheme 15 | ||
From the kinetic data they suggested that Cu(hfacac)2 (75) reversibly coordinates to the oxirane 74 lone pair of electrons to form the substrate–catalyst complex 78. Complex 78 then reacts with another Cu(hfacac)2 (75) and give a chelated substrate-bound cationic copper(II) complex 79. Here [Cu(hfacac)3]− serves as the non-nucleophilic counterion (X−).81 Acting as an optimal Lewis acid this cationic catalyst holds the substrate together in such a way that the chirality of C–O bond is transferred without scrambling to the olefin terminus and thereby forms the dihydrofuran–bound copper complex 80. As complex 80 is a weaker chelate than 79 it promptly coordinates to another vinyl oxirane 74 and releases the product 76 thereby reforming 79 and completing the first round of the catalytic cycle (Scheme 16).
 | ||
| Scheme 16 | ||
2.5. Cyclopropanation reactions
Salvileucalin B (83) is cytotoxic against A549 (human lung adenocarcinoma) and HT-29 (human colon adenocarcinoma) cells.82 Single crystal X-ray diffraction revealed that it is comprised of an unusual norcaradiene core embedded within the polycyclic carbon skeleton. Reisman et al. reported a synthetic strategy on the basis of a copper-catalyzed intramolecular arene cyclopropanation to afford the central norcaradiene core of Salvileucalin B. Retrosynthetic study reveals that salvlleucalin B could be prepared from the substrate 82. Access to 82 via cyclopropanation of 81 requires the chemoselective intramolecular reaction of a metal-carbenoid (derived from the R-diazo carbonyl of 81) with the arene π-system, in preference to highly favorable C–H insertion reactions at the adjacent, activated benzylic proton sites.83 Thus they prepared R-diazo ketone 81 and a number of metal catalysts were evaluated for their ability to promote the formation of norcaradiene 82. Though Rh(II)-catalyst favored the formation of C–H insertion products it was found that Cu(acac)2 provided more promising levels of cyclopropanation. After screening several Cu(II) catalysts it was concluded that relatively electron-poor Cu(II) salts provide the best results to yield norcaradiene 82. The microwave-promoted reaction proved useful only in the context of catalyst screening. However, improved yields were obtained for substrate 81 by using a slow addition protocol. Thus, syringe pump addition of 81 to a solution of Cu(hfacac)2 in dichloroethane at reflux cleanly provided the norcaradiene 82 in 73% isolated yield (Scheme 17).84 | ||
| Scheme 17 | ||
Lathyranoic acid A 86 was first isolated from the seeds of Euphorbia lathyris. It is a traditional Chinese medicine and used for the treatment of hydropsy, ascites, scabies, and snake bites.85 Lathyranoic acid A also shows various interesting biological properties including the activation of protein kinase C, anticancer and anti-HIV activity as well as P-glycoprotein inhibition.86 The structure of lathyranoic acid A possesses a stereo-defined cis-vinylcyclopropane and a 2-methylene-1,3-alkoxide. The interesting structural feature along with the potential biological activity attracted the attention of Nan et al. They reported the first total synthesis of lathyranoic acid A in a linear sequence of 20 steps and in 1.4% overall yield. This synthesis afforded a stereocontrolled Cu-catalyzed intramolecular cyclopropanation to construct the cis-cyclopropane unit as a key step. Copper-catalyzed intramolecular cyclopropanation of diazoacetate 84 afforded the exo intermediate bicyclic lactone 85 as a single product without the use of a chiral catalyst. The absolute configuration of 85 was confirmed by the single crystal XRD analysis of its deprotected derivative (Scheme 18).87
 | ||
| Scheme 18 | ||
The antibiotics, e.g., (−)-platencin 91 and (−)-platensimycin 92 were isolated from Streptomyces platensis MA 7327 and 7339, respectively.88 (−)-Platensimycin selectively inhibits FabF, the condensing enzyme, that catalyzes elongation in bacterial fatty acid synthesis. On the other hand, (−)-platencin can moderately inhibit both FabF and FabH, the enzyme catalyzing the initial condensation in bacterial fatty acid synthesis. Moreover, both 91 and 92 show potent, broad-spectrum Gram-positive antibacterial activity though they do not exhibit any cross-resistance to antibiotic-resistant bacteria, including MRSA and VRE.
Njardarson et al. developed a very efficient route to the compact platensimycin core 90 using a new copper-catalyzed oxirane ring expansion in combination with an alkylative dearomatization to complete the core.89 On the other hand, Nakada et al. recently accomplished90 a divergent approach to (−)-platencin and (−)-platensimycin via highly enantioselective catalytic asymmetric intramolecular cyclopropanation (CAIMCP) of α-diazo-β-keto sulfone developed in their laboratory. The CAIMCP reaction using CuOTf and ligand 88 proceeded at room temperature to successfully afford the cyclopropane 89 in 72% yield with 95% ee. It was also observed that the CAIMCP reaction with other dihydrooxazole ligands containing a bulky substituent proceeded slowly and required heating. The yields of the reactions are low perhaps owing to the crowded transition states that arose from the bulky substituent in the ligands. The intermediate 89 obtained by the CAIMCP of 87 is the common intermediate of both (−)-platensimycin and (−)-platencin (Scheme 19).
 | ||
| Scheme 19 | ||
2.6. Miscellaneous
The hydroxylated phenanthridones, isocarbostryls, belong to an interesting class of biologically active natural products of the Amaryllidaceae group.91 trans-Dihydronarciclasine (98) is one of the important members of this family that shows potent antitumor and antiviral activity. It also exhibits high activity against selected human cancer cell lines.92 Studer et al. reported the first total synthesis of enantiomerically pure 98 using a copper-catalyzed enantioselective nitroso Diels–Alder reaction for the transformation of racemic dienes 94 to the adduct 97 as a key step. The reaction of the diene 94 afforded the desired regioisomer 97 in 48% yield with excellent enantioselectivity (>99% ee) (Scheme 20).93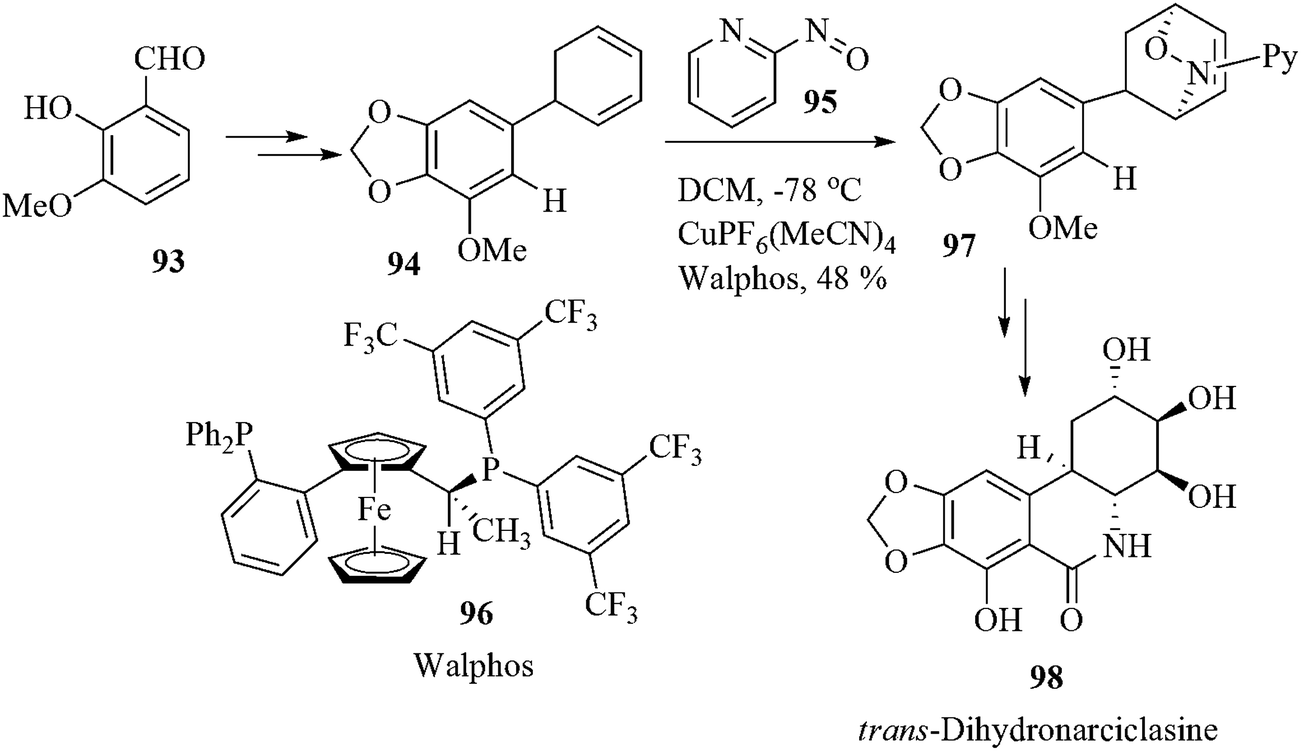 | ||
| Scheme 20 | ||
The first total synthesis of marine toxin (−)-gymnodimine 104 has been accomplished by a highly diastereo- and enantioselective exo Diels–Alder reaction using bis-oxazoline Cu(II) catalysts (copper-box).94 Gymnodimine is a special class of spirocyclic imine toxin containing a 6,6-spirocyclic imine as well as one butanolide appendage. Though it exhibits high toxicity this is reduced when administered with food.95 It shows toxic effect via binding to nicotinic acetylcholine receptors with picomolar affinities.96 The Kong group94 found that the reaction between lactam 99 and Z-diene 100 failed to give any cyclized product. Changing the chiral catalyst and δ-lactam activating group also proved unsuccessful. Perhaps the olefinic geometry is responsible for the lack of reactivity. They then prepared the asymmetric Diels–Alder precursor E-diene 101 and studied the reaction. Pleasingly, in the presence of Lewis acid [Cu (box)](SbF6)2, the E-diene 101 afforded the product 102 in excellent yield and with high diastereoselectivity (Scheme 21).
 | ||
| Scheme 21 | ||
Two new, highly oxygenated natural products polemannone B (107b) and polemannone C (107c) were isolated from the roots of Polemannia montana.97 Surprisingly, there have been no synthetic efforts directed towards the polemannones, nor have the polemannones been subjected to biological evaluation. Recently, Lindsley et al. developed a novel, catalytic CuCl2/(−)-sparteine oxidative β,β-phenolic coupling reaction of styrenyl phenols followed by a rapid inverse-electron demand Diels–Alder reaction to afford the benzoxanthanone natural product carpanone 107a in good yield and as a single diastereomer.98 This new methodology has proved to be general with regard to substrate scope, but not enantioselective. The first total synthesis of polemannones B and C employing a catalytic Cu(II)/(−)-sparteine oxidant system as a single diastereomer in overall yields of 15% and 31.5%, respectively, were also accomplished. (Scheme 22).99
 | ||
| Scheme 22 | ||
The marine natural product (−)-manzacidin B was diastereoselectively synthesized by a novel copper-catalyzed aldol reaction of α-methylserine-derived aldehyde with an isocyanoacetate possessing (1R)-camphorsultam. The reaction gave the (4R,5R,6R)-adduct, which after a few steps was converted into manzacidin B.100 Manzacidin B (112) was isolated from the Okinawan sponge Hymeniacidon sp. and its structure consists of an ester-linked bromopyrrolecarboxylic acid and a 3,4,5,6-tetrahydropyrimidine ring in which one of the amino groups is attached to the C6 quaternary carbon center.101
It was observed that the reaction of the ring-opened aldehyde 108 with (1R)-109 showed the (4R,5R)-selectivity to give the desired (4R,5R)-110 as the major isomer (59%, 110/111 = 13:1). Using the MOM-protected 108 with (1R)-109 was found to be the best combination to obtain (4R,5R)-110 in view of the yield and diastereoselectivity (84%, dr = 13:1). Inspite of the mismatched pair between 108 and (1R)-camphorsultam 109, the chiral auxiliary based diastereocontrol was ascribed to the high diastereoselective formation of the desired (4R,5R)-110 (13:1). Scheme 23 depicts the proposed transition state model of the chiral camphorsultam aldol reaction to give 110. Preferentially the Z-enolate would be formed102 in order to avoid steric and/or electronic repulsions between the isocyanide and sulfone groups. In this model it was assumed that approaches from the re-face of the Z-enolate of (1R)-109 and the re-face of the aldehyde 108 would be the kinetically favored process to give (4R,5R)-110 as the major diastereomer.
 | ||
| Scheme 23 | ||
The first total synthesis of Resolvin D6 113 has been achieved by using a mild copper-catalyzed coupling of cis-1,4-dibromo-2-butene with TMS-acetylene as one of the key steps (Fig. 6).103 It is an endogenous lipid mediator of resolution of inflammation derived from docosahexaenoic acid.
 | ||
| Fig. 6 Resolvin D6 | ||
3. Silver-catalyzed synthesis of natural products
Recently, the development and application of silver-based methodologies have proven to be very useful in the synthesis of simple to complex molecules including natural product synthesis. The silver-catalyzed/mediated transformations are increasingly used in cycloadditions, allylations of carbonyl and imine groups, aldol reactions etc. Intramolecular heterocyclizations, C–H bond activation of terminal or silylated alkynes utilizes its asymmetric version using chiral ligands. The σ- and/or π-Lewis acidity of Ag(I) can activate the substrate and the substrate activation often converts its transition metal partners into “hotter” cationic complexes.3.1. Cycloaddition reactions
The cyanocyclines, SF-1739HP, dnacins (Actinosynnema pretiosum C-14482), bioxalomycins (Streptomyces viridostaticus and S. lusitanus), and aclidinomycins (Streptomyces halstedii KB012) cover the naphthyridinomycin family of tetrahydroisoquinoline antibiotics.104 These natural products share a number of structural features, mainly their ABCD ring skeleta, with the simpler quinocarcin family of tetrahydroisoquinolines and also exhibit varying degrees of antibacterial and anticancer activities. Most of them interact with DNA. Cyanocycline A 119, one of the most important members of the naphthyridinomycin family, has been synthesized.105 There are several reports regarding the total synthesis of these natural products. The Evans, Illig and Fukuyama groups independently synthesized the racemic cyanocycline A in 30 steps from cyclopentadiene, 35 steps from 2,6-dimethoxytoluene and 32 steps from tert-butyl azidoformate, respectively.106The Garner group107 used the silver-catalyzed asymmetric [C + NC + CC] coupling reaction as a key step in the synthesis of cyanocycline A. The aldehyde 115, prepared from the serinal derivative 114, underwent the essential asymmetric [C + NC + CC] coupling reaction with 1.1 equiv. glycylsultam 116, 3 equiv. dipolarophile, and 5 mol% AgOAc to afford a mixture of pyrrolidine 117 and its isomer in combined 38% yield along with a mixture of imidazolines and oxazolidines. Formation of these byproducts is indicative of ineffective trapping of the sterically congested azomethine ylide by a relatively poor dipolarophile. This problem was solved by optimization of the reaction conditions. The reaction was found to be successful when methyl acrylate was used as a solvent and the amount of catalyst was increased to 10 mol%. The desired pyrrolidine was poduced in 73% isolated yield. It is assumed that the [3 + 2] cycloaddition proceeds via a pre-TS ensemble such as 118. The diastereofacial selectivity in this model is determined by an ylide conformation that places the N-acyl sultam CO and SO2 dipoles anti to each other while still maintaining the usual imine-carbonyl chelation by Ag(I). In this model, the coordinated acrylate dipolarophile approaches the ylide from the least hindered endo-si face opposite to the pro-R sulfoxide moiety. This synthetic procedure offers cyanocycline A in 22 linear steps from 2,6-dimethoxytoluene (19 steps from the readily available serinal 114) and cyanocycline A can easily be converted to bioxalomycin β2 through the agency of Ag(I) (Scheme 24).
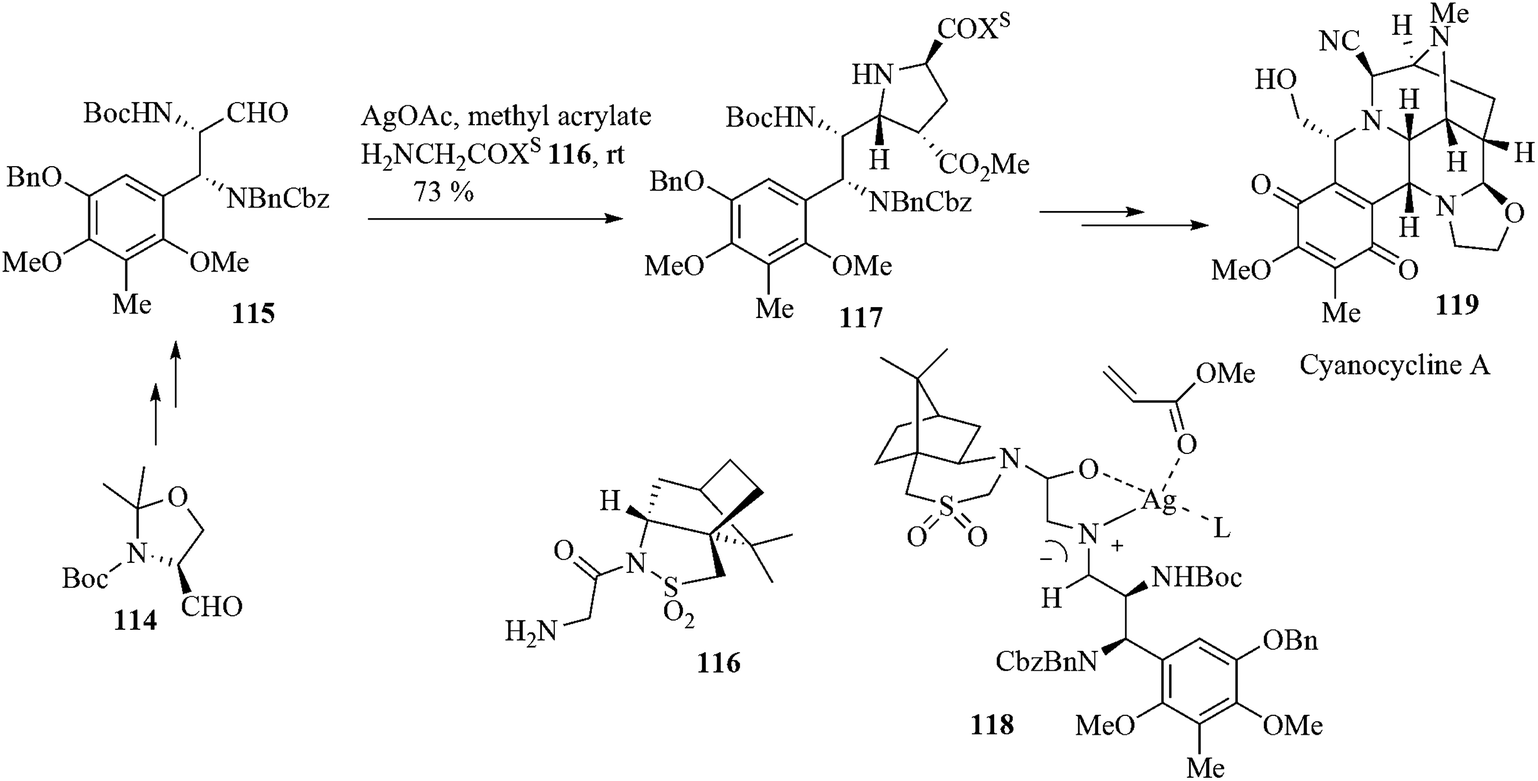 | ||
| Scheme 24 | ||
The crinine-type alkaloids were isolated from plants of the Amaryllidaceae108 family which has produced huge number of structurally diverse alkaloids possessing a wide range of interesting physiological effects including antitumor, antiviral, acetylcholinesterase inhibitory, immunostimulatory and antimalarial activities.109 Among them maritidine 123b and its structural analogues were isolated from the Pancratium maritimum, Pancratium tortuosum, and Zephyranthes genera.110 They possess a 5,10b-ethanophenanthridine nucleus containing dimethoxy substituents at the C-8 and C-9 positions.
The Pandey group accomplished the construction of both C4a–C10b and C11–C12 bonds of 122 in one step with the required stereochemistry via the intramolecular [3 + 2]-cycloaddition reaction of non-stabilized azomethine ylide containing a tethered geminally disubstituted dipolarophile. Silver(I) fluoride was used as a one-electron oxidant to generate the corresponding azomethine ylide intermediate in situ from the corresponding starting material 120. Thus the cycloaddition reaction was performed by dropwise addition of 120 in dichloromethane to a stirring mixture of flame-dried silver(I) fluoride in anhydrous dichloromethane. The reaction afforded the desired cycloadduct 122 in 56% isolated yield (Scheme 25).111
 | ||
| Scheme 25 | ||
The first total synthesis of natural product sorocenol B (124) in racemic form was achieved by Porco et al.112 The retrosynthetic analysis reveals that compound 124 may be derived from MOM acetal precursor 125 which may be prepared by the application of AgNP-catalyzed Diels–Alder cycloaddition113 between hydroxychalcone 126 and diene 127. The compounds 126 and 127 were derived from commercially available chromene 128 and resorcinol 129, respectively (Scheme 26).
 | ||
| Scheme 26 | ||
They employed a highly efficient and user-friendly silver nanoparticle (AgNP)-catalyzed Diels–Alder cycloaddition114 reaction and at a late-stage Pd(II)-catalyzed oxidative cyclization as key steps. Sorocenol B possesses an intriguing bicyclo[3.3.1] core and was first isolated from the root bark of Sorocea bonplandii.115 It exhibits antitumor activity with a mean cell growth of 26% of control and subsequent full-dose response assays also showed low micromolar level cytotoxic activities and moderate selectivity. The chalcone 126 and diene 127 were used as the key starting materials for a silica-supported silver nanoparticles (AgNP's)-catalyzed Diels–Alder cycloaddition reaction. Only 0.1 mol% catalyst loading afforded the desired cycloadducts in 90% combined yield with a 2:1 ratio of separable endo/exo diastereomers. The catalyst-free thermal Diels–Alder cycloaddition afforded a very low yield of the cycloaddition product (Scheme 27).
 | ||
| Scheme 27 | ||
The reaction is initiated by the proton removal and single electron transfer (SET) from the adsorbed chalcone 126 to the AgNP.116 This generates the AgNP-stabilized phenoxy radical intermediate 130a which is in resonance with carbon-centered radical 130b. Concerted [4 + 2] cycloaddition between the activated dienophile 130a/b and diene 127 provides intermediate 131. Intermediate 131 generates 132 via back electron transfer (BET) and protonation. A final desorption step turns over the catalytic cycle and releases cycloadduct 125 into solution. During the entire reaction the AgNP's thus serve as an “electron shuttle” catalyst117 leading to highly selective activation of 2′-hydroxychalcones for the cycloaddition. Interestingly, no cycloadduct was formed when 2′-methoxychalcone was used as a dienophile, indicating the requirement of 2′-hydroxyl substituent in the chalcone substrate (Scheme 28).
 | ||
| Scheme 28 | ||
Biologically active and architecturally unique secondary metabolite (−)-berkelic acid 139 was isolated from an extremophilic Penicillium species obtained from the surface water of Berkeley Pit Lake in Butte, USA.118 The literature reveals that it can inhibit the cysteine protease caspase-1 (98 mm), matrix metalloproteinase MMP-3 (1.87 mm) and displays selective activity against the human ovarian cancer line OVCAR-3 (GI50 = 91 nM). Though three research groups accomplished the total synthesis of 139 the strategies used afforded very low yields of the natural product119 that was insufficient to carry out bioactivity studies. Owing to this, comparative results were contradictory.120 The de Brabander group121 in their total synthesis of berkelic acid designed a strategy in such a way where in situ dearomatization of lactol 138 to pulvilloric acid methyl ester formation (compound 137) as well as tandem C–C bond formation with spicifernin-like fragment 135 could take place simultaneously. In this respect the Ag+ was the best choice due to its proper balance of hard Lewis acidic properties and alkynophilic character. The former removes ethanol from 138 and the latter induces cycloisomerization of the alkynol 135 to enol ether 136.122 The lactol 138 and AgSbF6 (3.5 equiv.) in the presence of alkynols 134, 135 resulted in the formation of methyl berkelate which after (Bu3Sn)2O-catalyzed deprotection afforded the berkelic acid 139. This route offered a highly convergent and efficient synthesis of berkelic acid and the stereochemistry at C 22 was also established (Scheme 29).
 | ||
| Scheme 29 | ||
Recently, Rodriguez et al. undertook the challenge and accomplished the total synthesis of berkelic acid wherein all but the last step were executed on the gram scale. This level of practicality enabled a highly stereoconvergent and modular synthetic strategy.123 The group used their newly developed palladium(II)-catalyzed one pot three-component coupling reaction for the diastereoselective construction of the chroman spiroacetal which is the key step of the total synthesis.124 The main essence of the reaction is the in situ formation of exocyclic enol ether 142 and a o-quinonemethide 137 as well as the subsequent formal cycloaddition reaction between these two reagents to produce chroman spiroacetal 143a. It was hypothesized that a metal complex could promote the cycloisomerization of the alkyndiol as well as the cycloisomerization of the aldehyde would give 137. In fact the reaction was found to proceed smoothly in the presence of 5 mol% of AgOTf. The possible decomposition of the synthesized compound in the presence of the reactive newly formed pyran ring could be avoided by direct hydrogenation of the carbon–carbon double bond of the pyran ring under conventional conditions. This produced the reduced intermediate 143b. Notably, four new chiral centers were formed in this transformation and only two diastereoisomers were obtained in the crude reaction mixture (dr = 2:1). The major diastereoisomer was found to be the desired product 143 with all chiral centers and functionalities resembling those of the natural product. It is remarkable that this reaction could be performed on a gram scale in 83% yield. This new silver-catalyzed reaction allowed the assembly of the central core of the natural product possessing four rings and five stereocenters in only one step (Scheme 30).
 | ||
| Scheme 30 | ||
3.2. Alkyne/allene–hydration reactions
Amphidinolides are biologically active macrolides, isolated from the dinoflagellate Amphidinium sp.125 Among them amphidinolide F 147 contains eleven stereogenic centers embedded within a 25-membered macrolactone including two trans-disposed tetrahydrofuran ring systems and possesses significant cytotoxic activity.126 Recently, Carter et al. disclosed the first total synthesis of amphidinolide F and established both the absolute and relative stereochemistry of the natural product.127Initially, a gold-catalyzed cyclization of 144 based on the work of Gagosz128 and Krause129 was employed but without any success. When the use of AgBF4 was attempted instead of gold pleasingly the desired dihydrofuran 146 was obtained in good yield and with complete stereoselectivity. This transformation was found to be effective in 5 gram scale and provided sufficient quantities of 146, which might serve as a building block for the synthesis of a variety of trans-disposed furan-containing natural products (Scheme 31).
 | ||
| Scheme 31 | ||
Cassiarin A 151 and cassiarin B 152 were isolated from the leaves of Cassia siamea and are used in traditional medicine particularly for the treatment of periodic fever and malaria.130 They also exhibit potent antiplasmodial activity against Plasmodium falciparum with an IC50 0.005 and 6.9 µg mL−1, respectively. Yao et al. accomplished the total synthesis of cassiarins A and B using a Ag(I)-promoted formation of the tricyclic 8H-pyrano[2,3,4-de]chromen-8-one core as the key step. The synthetic strategy was accomplished either via salt intermediate 150 or neutral intermediate 149 (Scheme 32).131
 | ||
| Scheme 32 | ||
3.3. Carbon–nitrogen coupling reactions
Microcin B17 (MccB17) 157, secreted from various strains of the bacterium Escherichia coli,132 is a 43 amino acid antibacterial peptide. It targets DNA gyrase, a member of the type II topoisomerase family of enzymes, essential for DNA replication in prokaryotic organisms.133 It also shows extensive post-translational modification, via enzymatic dehydrative cyclization and dehydrogenation of specific serine and cysteine residues to give several thiazole and oxazole heterocycles throughout the native peptide backbone.134 Payne et al. undertook the total synthesis of MccB17 which includs the use of Fmoc-SPPS to rapidly assemble three key peptide and peptide thioester fragments under Ag(I)-mediated ligation condition.The disconnection of MccB17 was proposed at two junctions, namely Gly19–Gly20 and Gly34–Gly35. They individually synthesized target peptide 153 and peptide thioesters 154 and 156 that could be obtained through Fmoc-strategy SPPS.135 Initially, they performed the direct aminolysis ligation reaction between the peptide 153 and peptide thioester 154 using Wong's methodology (4:1 v/v NMP/Gn.HCl, HEPES, pH 7.5, thiophenol). The reaction was carried out for 7 days followed by treatment with aqueous hydrazine and afforded the desired product MccB17 155 in only 38% yield. This observation required an alternative coupling strategy. The use of a Ag(I)-promoted ligation reaction was possible due to the presence of a nonepimerizable C-terminal glycine residue.136 The coupling of 153 and 154 in the presence of AgNO3, 3-hydroxy-1,2,3-benzotriazin-4(3H)-one (HOOBt), and N,N-diisopropylethylamine in DMSO for 18 h furnished the intermediate 155 in 70% yield. The same Ag-catalyzed coupling strategy was employed for the crucial final ligation. The peptide 155 and thioester 156 under the reaction condition (AgNO3, HOOBt, iPr2NEt, DMSO) afforded the desired compound 157 after 24 h in 45% yield over two steps (Scheme 33).137
 | ||
| Scheme 33 | ||
3.4. Carbon–carbon coupling reactions
The construction of 3,4-disubstituted pyrrole systems as well as selective substitutions at one or more of the β-positions has been a challenging goal in many of the synthetic programs. Moreover, functionalization of pyrrole N systems is difficult due to lack of selectivity and significant polymerization.138 In 2010, Jia et al. utilized a one pot AgOAc-mediated oxidative coupling reaction for the synthesis of 1,3,4-trisubstituted pyrroles and successfully applied this to the synthesis of purpurone 165, a potent ATP-citrate lyase (ACL) inhibitor.139 The purpurone 165 belongs to some important marine natural products.140 The suggested mechanism of the pyrrole forming reaction involves the following steps: (i) the imine formation between the amine 158 and aldehyde 159 (ii) isomerization of 160 to produce substituted enamine 161 (iii) oxidation of 161 to produce radical cation 162 (iv) intermolecular coupling to produce the dimeric salt 163 and finally (v) elimination of one aniline derivative 158 to produce the desired pyrrole 164. The key intermediate of purpurone (165) was obtained in 59% yield (Scheme 34).141 | ||
| Scheme 34 | ||
Pentacyclic tetrahydroisoquinoline alkaloid (−)-quinocarcin 168 was isolated from the culture broth of Streptomyces melun nucleus,142 which possesses potent antitumor activities against a variety of tumor cell lines. Under anaerobic conditions quinocarcin underwent self-redox disproportionation to produce inactive quinocarcinol and quinocarcinamide. Its fascinating molecular architecture and important biological profile attracted significant attention from the synthetic community resulting one racemic143 and three asymmetric synthesis of (−)-quinocarcin.144 In 2008, Zhu et al.145 reported the synthesis of (−)-quinocarcin in the longest linear sequence of 22 steps from 3-hydroxybenzaldehyde in 16% overall yield.
A careful disconnection study revealed that the labile oxazolidine ring could be installed at a late stage of the synthesis to construct the strained 3,8-diazabicyclo [3.2.1] octane ring. It is assumed that the reaction proceeds through the N-acyliminium intermediate 169. But practically the compound 169 was converted to the stable intermediate 166 in the presence of strong external nucleophile EtSH. The synthesis was accomplished via a silver-promoted intramolecular Mannich reaction using silyl enol ether as a nucleophile as one of the key steps. The intramolecular Mannich reaction of 166 was performed in the presence of silver tetrafluoroborate in THF at rt. The exo-oriented tetra cyclic aldehyde 167 was isolated in 88% yield. Silver tetrafluoroborate acted as an activator of both electrophile and nucleophile in this cyclization leading to a disfavored 5-endo-trig cyclization (Scheme 35).146
 | ||
| Scheme 35 | ||
3.5. Cascade reactions
Several insect sex pheromones, polyketide antibiotics and microtubule stabilizing agents consist of a common structural component, namely the spiroacetals.147 Moreover, the 5,5-spiroacetal-containing fungal metabolites demonstrate potentially useful biological activities.148 Among them cephalosporolides, penisporolides, and ascospiroketals exhibit anti-inflammatory activities.149 Britton et al. examined150 the Ag-promoted cyclization of 171 based on their earlier reported microwave-assisted preparation of tetrahydrofuranols from chloropolyols strategy.151 A 1:1 mixture of the desired epimeric spiroacetals 172a and 172b in overall 75% yield was obtained by the reaction of the ketochlorohydrin with AgOTf-Ag2O catalytic system. Interestingly, Ag2O played a crucial role by converting the trifluoromethanesulfonic acid (TfOH) generated in the reaction into AgOTf, thus effectively buffering the reaction media. The effective conversion of TfOH into AgOTf was assumed to occur on the surface of largely insoluble Ag(I) particles. So using sonication the reaction time was lowered and the overall yield of the reaction was increased. The surface area and also buffering capacity of the Ag2O were increased by sonication. Finally, the pendant alkene was cleaved under oxidative condition to provide cephalosporolide E 173a and cephalosporolide F 173b (Scheme 36).
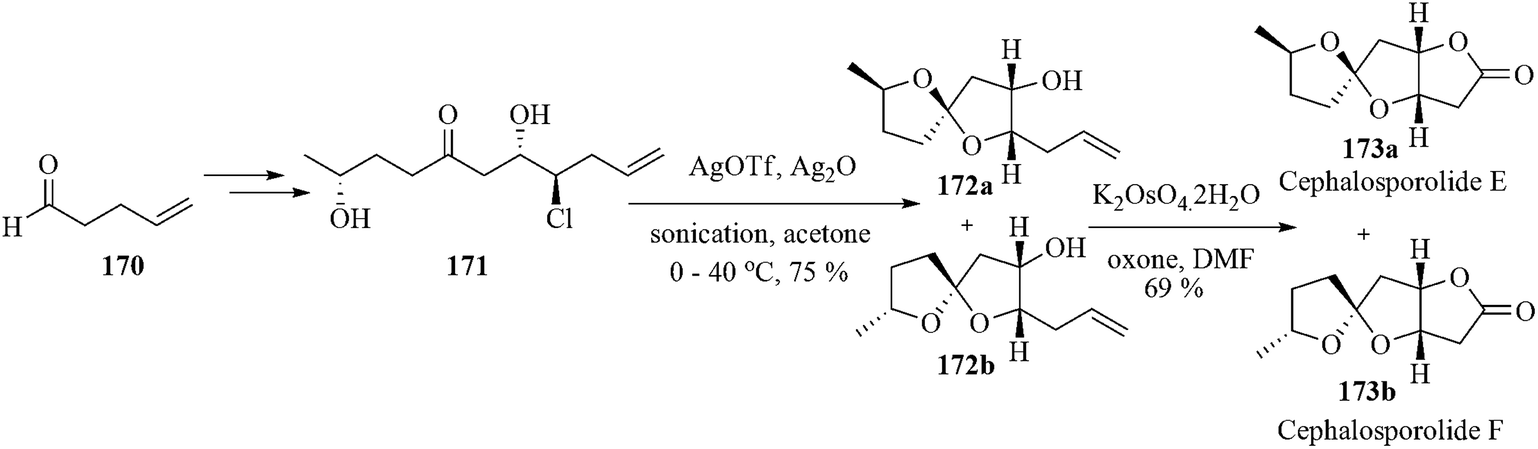 | ||
| Scheme 36 | ||
A silver(I)-initiated hydroamination cascade reaction to construct the bicyclic guanidinium ion core from alkynyl bisguanidine was employed for a concise stereoselective total synthesis of (+)-saxitoxin 178. This is a one pot reaction strategy that produces a single stereoisomer in a single synthetic transformation sequence via two CN bonds, one CO bond and three rings formation. The siignificant toxicity of saxitoxin has led to its use as an ion channel probe and chemical warfare agent.152 Synthetically it is a challenging target due to its very polar, heteroatom rich, and densely-functionalized molecular architecture.
Initially, a futile attempt was made to form the bicyclic guanidine oxidatively from 174 via epoxidation with DMDO or mCPBA. The alternative was to use an electrophilic halogen source to trigger the formation of the C4 aminal. The reaction placed a halogen at C12 instead of an oxygen atom in a proper position to be displaced by an adjacent Boc group suggestive of the Woodward dihydroxylation.153 This strategic change differentiates the two guanidines required to successfully execute the final annulation sequence as well as to afford the bicyclic aminal 176 as a single diastereomer. Treatment of the alkyl iodide 176 with Ag(I) and AcOH yielded the oxazolidinone 177 in which only a single Boc group had participated.154 On the other hand, the tricyclic carbamate 177 is supposed to be formed by using a one pot Ag(I)-catalyzed process via 174 → 175 → 176 → 177. The intermediate 174 was converted to 177 in good isolated yield. This involved the formation of two CN bonds, one CO bond, and three rings from an acyclic precursor in a single synthetic manipulation (Scheme 37).155
 | ||
| Scheme 37 | ||
Waldmann et al. reported156 a silver-catalyzed microwave-assisted one-pot cascade synthesis to access diverse alkaloid-inspired scaffolds including a concise total synthesis of fascaplysin 185 and homofascaplysin C 186. The fundamental process of the strategy is an imine formation from acetylenic benzaldehyde and aniline possessing a pendant nucleophile. The nucleophile subsequently undergoes an intramolecular cycloisomerization in the presence of Ag/Au ions as catalysts that activate the alkyne for nucleophilic attack to yield the isoquinolinium intermediate.
Initially, the boc-protected 3-ethynyl-indole-2-carbaldehyde 179 was used as a common precursor for the target molecule fascaplysin and homofascaplysin C. The pentacyclic core 181 was obtained in high yield by the microwave-assisted silver-catalyzed cascade cyclization of 179 with anilines 180 and followed by acidic work-up. It was observed that individual use of AuCl3 or AgOTf in dichloroethane or ethanol afforded only a relatively low yield of 181 whereas a combination of AuCl3 with a silver co-catalyst in dichloroethane resulted in a higher yield. However, the yield was significantly increased when 20 mol% of AgOTf was used at 60 °C in ethanol along with an excess of a nitrogen base like 2,6-lutidine (Scheme 38).
 | ||
| Scheme 38 | ||
3.6. Miscellaneous
Qin et al. achieved the total synthesis of (−)-acetylardeemin, (−)-ardeemin and (−)-formylardeemin starting from a common precursor using silver-promoted direct Friedel–Crafts alkylation as a key step (Fig. 7).157 (−)-Acetylardeemin 187a and (−)-ardeemin 187b were isolated from the extracts of the fungus Aspergillus fischeri which are the most potent inhibitors of multidrug resistance.158 On the other hand, (−)-formylardeemin 187c at low µM doses significantly sensitizes the multi-drug resistant breast cancer cell lines MCF-7-R and cervical cancer cell lines Siha to vincristine- or adriamycin-induced cell death.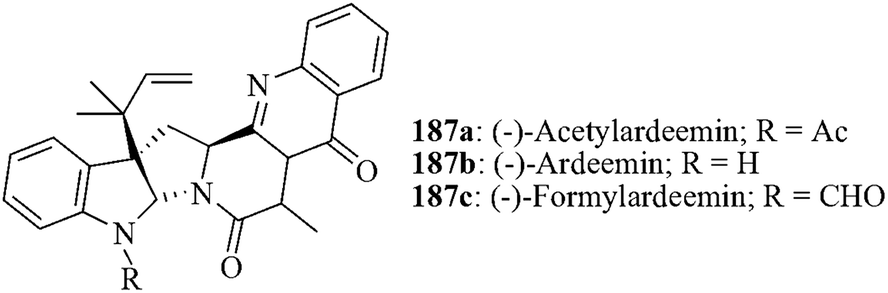 | ||
| Fig. 7 Pyrroloindoline alkaloids | ||
4. Gold-catalyzed synthesis of natural products
Gold catalysis in the synthesis of organic compounds has truly expanded in just a few years. Ag(I) and Au(I) exhibit strong Lewis acidity (i.e., is π-electrophilic) toward unsaturated C–C bonds, as well as toward lone pairs of electrons associated with heteroatoms (i.e., is σ-electrophilic). The gold catalysts are stable in air and moisture and the reaction can be carried out under milder conditions.159 Different useful transformations including additions of N, O, S, and C nucleophiles to C–C unsaturated bonds; Friedel–Crafts type reactions; activation of carbonyl and imine groups; cycloisomerizations of enynes and heteroenynes; and activation of terminal C–H bonds, can be efficiently accomplished using gold catalysis. Asymmetric catalysis by nonracemic Au complexes is also an area of recent research interest. In particular, the gold-catalyzed cycloisomerization of alkynol-based systems has been used for the synthesis of different heterocyclic structures e.g., furans, dihydrofurans, pyrans, furanones and others including naturally occurring structures.1604.1. Alkyne-hydration reactions
Hydration of alkyne is a valuable transformation in the context of chemical synthesis. The electrophilic mercury salt can easily convert alkynes into ketones though the toxicity of mercury suppresses their wide use. In this respect alternative transition-metal catalysts have been developed, with gold complexes taking a leading role. The gold-catalyzed addition of an alcohol or carboxylic acid across a triple bond is very much useful as it can prevent unwanted side reactions and offer good conversions to provide valuable routes to sensitive and complex natural products. Forsyth, Trost, Furstner, Dudley, and Floreancig groups frequently applied the gold-catalyzed route to a variety of complex natural products.161 Stout et al. extracted a polyunsaturated α-pyrone derivative, dubbed neurymenolide A 198, by the bioassay-guided fractionations of the red alga Neurymenia fraxinifolia harvested off Fiji island. This metabolite is very much active against methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus faecium (VREF), and shows moderate cytotoxicity against 12 human cancer cell lines.162It was previously reported that the substrate 188 can easily be transformed into the corresponding pyrone derivative 189 by treatment with 1 mol% of [(SPhos)AuNTf2] (190; Tf = trifluoromethanesulfonate) at ambient temperature.163 Initially the active gold complex coordinates to the triple bond and subsequent 6-endo dig cyclization produces the intermediate 192. The release of substituted alkene (through cleavage of the tert-butyl group) and subsequent protonation as well as acetylation produces the pyrone derivative 189. Based on this observation the Furstner group undertook the total synthesis of neurymenolide A. Rapid decomposition occurred when 195 was treated with a catalytic amount of 190 in nitromethane (aprotic solvent). After detailed experimentation it was found that the gold complex 196, containing a bulky Xphos ligand, allowed the formation of the desired product 197 in modest (26%) yield along with unreacted starting material (37%). Use of HOAc as a co-solvent was found to be better. It accelerated the critical proto-deauration step within the catalytic cycle, resulting in a much cleaner transformation. The pyrone 197 was isolated in 73% yield after acetylation of the crude material. Thus the fast isomerization of the lateral alkene was suppressed by this in situ protection. The selective activation of the least electron-rich acetylene group in 195 is kinetic in origin and led to pyrone formation. The reaction is irreversible due to the gain of aromaticity (Scheme 39).164
 | ||
| Scheme 39 | ||
Naturally occurring antibiotic abyssomicin C 206 was isolated in 2004 by Sussmuth and co-workers.165 It inhibits tetrahydrofolate biosynthesis at an early stage.166 There is no precedence for the mechanism of antibiotic activity of abyssomicin C against Gram-positive bacteria, including methycillin- and vancomycin-resistant Staphylococcus aureus strains. Due to its strong antibacterial activity as well as complex molecular architecture, several groups considered abyssomicin C as an attractive synthetic target.167 All the research groups individually constructed the central cyclohexane core of abyssomicin C 206 by a Diels–Alder reaction. Saicic et al. recently developed an enantioselective synthesis of atrop-abyssomicin C based on a gold-catalyzed alkyne hydration strategy as the key cyclization step.168
Initially, they synthesized compound 199 as an alkyne-hydration precursor and applied nafion-Hg-mediated cyclization strategy upon it. 199 underwent cyclization to give spirotetronate 200, but failed to convert 200 into tricyclic ether 202. It was realized that an extremely unfavorable distorted conformation of the rigid spirobicycle is required in the transition state 201 for the intramolecular hetero-Michael addition (i.e. to allow the approach of the hydroxy group to the alkene acceptor at a Burgi–Dunitz angle of 105°). Therefore, an alternative protocol was designed, via the intramolecular etherification on a conformationally more flexible alkyne derivative, prior to spirotetronate formation.
The best alternative was found to be gold complexes,169 efficient promoters of nucleophilic additions to carbon–carbon multiple bonds. No reaction occurred whether the alkyne 203 was treated with AuCl3, in the presence or absence of silver triflate. Therefore, a highly active Au(I) catalyst was utilized. When a solution of the alkyne 203 in THF was treated with Gagosz's gold catalyst170 no reaction occurred, but a dichloromethane solution of the mixture on treatment with Gagosz's catalyst at 120 °C afforded the bicyclic ether 202. An optimization study of the reaction condition revealed that the intramolecular etherification could be effected by heating a solution of the alkyne 203 and a catalytic amount of [(PPh3)AuNTf2] in isopropanol. The reaction produced the Z-isomer of 205, (Z-205), while the key intermediate required for the synthesis of spirotetronate 202 is E isomer of 205, (E-205). Isomerization of (Z)-205 occurred by irradiation with UV light in a quartz vessel. The corresponding E-isomer on treatment with sodium hydride was cyclized to give the spirotetronate. Further experimentation showed that this three-step transformation could be accomplished more efficiently as a one-pot sequence. Heating the alkyne 203 in the presence of Gagosz's gold catalyst, followed by irradiation in the presence of a catalytic amount of sodium isopropoxide was found to be better. Initially, the (Z)-205 predominated in the photostationary state and subsequently this was converted to the (E)-205 that reacted to afford the tetronate 202. Thus it shifted the Z/E equilibrium. The principle of microscopic reversibility eliminates the separation of isomers and recycling and allows the formation of tricyclic tetronate 202 in a 60% overall yield (Scheme 40).
 | ||
| Scheme 40 | ||
The xyloketals are the ketal compounds having a cis disposition of three contiguous stereogenic centers embedded in the tetrahydrofuranobenzopyran moiety.171 Among them the xyloketal A 214a possesses a C3 symmetry. The xyloketal D 214b and its regioisomer xyloketal G 214c represent simpler structural analogues of xyloketal A.172 Xyloketal D inhibits acetylcholine esterase and is a potential lead compound for the treatment of neurological disorders like Alzheimer's disease.
Very recently, Sarkar et al. reported a gold-catalyzed xyloketalization reaction for the tandem synthesis of xyloketal natural products, xyloketal D and xyloketal G. The xyloketalization of 207 took place smoothly under the conditions AuCl3/TBAF/PPTS/MeOH/rt in near-quantitative yields.173 The reaction is initiated by the TBAF-catalyzed deprotection of the phenol silyl ether 207 to produce phenolic compound 209. The alkynyl complex 210 initiates the cycloisomerization reaction via the complexation of the triple bond. The coordination of the triple bond enhances the electrophilicity of the alkyne and the addition of alcohol occurs through a 5-exo-dig cyclization giving the vinyl gold intermediate 211. The proto-deauration and subsequent nucleophilic attack of the phenolic OH group furnishes the tricyclic ketal 208. Here PPTS acts as a proton source to yield 209 from the corresponding phenolate. It also promotes proto-deauration followed by oxonium ion formation for the conversion of 211 to 213 (Scheme 41).
 | ||
| Scheme 41 | ||
Polycavernoside A 218 is a glycosylated macrolide of the polycavernoside series. It possesses intricate structural features and significant biological activities.174 The Furstner group disclosed a concise total synthesis of this challenging marine natural product by a gold-catalyzed, highly regioselective, transannular hydroalkoxylation reaction.175
The cycloalkyne 215 was synthesized and subjected to transannular hydroalkoxylation with a view to forming the enol ether 217. It was previously reported that a soluble platinum catalyst acts well in this type of transannular hydroalkoxylation reaction.176 When the reaction was carried out in the presence of soluble PtCl2(C2H4)2 the alkyne was selectively attacked at the C9 position by the secondary OH group. But the reaction suffered from in situ hydrolysis of the intermediate enol ether to form the corresponding hemiketal. Simultaneously, the allylic ester was rearranged. However, the desired enol ether formation was accomplished in a good yield by using the cationic gold complex 216 bearing a bulky phosphine ligand. The compound 215 upon treatment with 216 in DCM afforded the intermediate 217, the key intermediate of polycavernoside A 218 in 84% yield (Scheme 42).
 | ||
| Scheme 42 | ||
Very recently, a formal total synthesis of didemniserinolipid B 219 was reported by Ramana et al. A regioselective gold-mediated 6-endo-dig alkynol-cycloisomerization reaction has been employed as a key step (Fig. 8).177
 | ||
| Fig. 8 Didemniserinolipid B | ||
4.2. Alkyne-amination reactions
Terreusinone (223) is a dipyrrolobenzoquinone containing a pyrrolo[2,3-f]indole-4,8-dione ring system that is unique among natural products. This was isolated from the algicolous marine fungus Aspergillus terreus.178 It exhibits significant UV-A protecting properties and thus protects the host organism from the harmful effects of solar UV radiation.179 Significantly, the compound 223 is the stronger UV-A protecting agent (ED50 ≈ 200 µm) than that of oxybenzone (ED50 = 350 µm), a compound widely used in sunscreens, suggesting that 223 may have potential dermatological and biomedical applications.Sperry et al.180 used the gold-catalyzed hydroamination of unsubstituted o-alkynylanilines to construct indoles. Initial attempts to achieve the desired transformation of 221 to 222 using all the reported gold(III) and gold(I) catalysts failed, primarily owing to the instability of the substrate 221. Echavarren's cationic gold(I) complex181 (acetonitrile)-[(2-biphenyl)di-tert-butylphosphine]gold(I) hexafluoroantimonate 216, was finally used. The pyrroloindole 222 was obtained in excellent isolated yield from 221 on exposure of 221 to 12 mol% of complex 216 in toluene at 60 °C for 30 min. This is the first reported synthesis of the photoprotecting dipyrrolobenzoquinone natural product (+)-terreusinone (Scheme 43).
 | ||
| Scheme 43 | ||
Recently, the Ohno group182 reported an efficient strategy using a gold(I)-catalyzed intramolecular alkyne hydroamination, as a simple and atom-economical protocol for the construction of the quinocarcin core structure. Initially, the hydroamination of 224 resulted in substantial decomposition using either catalyst 226 or 227. An increase of catalyst loading was also of no avail. Serious steric repulsion between the methyl ester and Boc groups that may impair formation of the required conformer for the hydroamination was then considered. So the corresponding amine 224b was prepared by cleavage of the Boc group. The desired 6-endo-dig cyclization proceeded efficiently on treatment of 224b with the catalyst 227. As the resulting enamine product was unstable, the desired 6-endo-dig product in the tetrahydroisoquinoline form was isolated after stereoselective reduction with NaBH3CN (Scheme 44).
 | ||
| Scheme 44 | ||
An approach to the total synthesis of piperidine alkaloids (+)-241D 229, isosolenopsin 230 and isosolenopsin A 231 was accomplished via a gold-mediated cyclization (Fig. 9).183
 | ||
| Fig. 9 Examples of naturally occurring piperidine alkaloids | ||
4.3. Miscellaneous
Most of the starfish saponins can be classified into two structural groups, the asterosaponins and the glycosides of polyhydroxysteroids. Among them the asterosaponins possess a Δ9(11)-3β,6α-dihydroxysteroidal nucleus along with a sulfate residue at C3 and a glycan at C6.184 The common glycans are penta- or hexasaccharides with all 1,2-trans-pyranosidic linkages and a (1 → 2) branching point at the second sugar unit. Goniopectenoside B 235 is one of the important members which was isolated from Goniopecten demonstrans, a large pinkish starfish living in deep waters of the Gulf of Mexico.185 The scarcest congener goniopectenoside B differs from other asterosaponins only at the side chain but shows similar antifouling activities.Yu et al. achieved the total synthesis of goniopectenoside B with a convergent linear sequence of only 21 steps and in 4.3% overall yield from adrenosterone. A novel gold-catalyzed coupling reaction between 232 and 233 produced the key intermediate 234 for the total synthesis of goniopectenoside B.186 They synthesized goniopectenoside B in a stereoselective manner. The coupling reaction of aglycon derivative 233 with pentasaccharide donor 232 bearing the neighbouring participating benzoyl group at C2, afforded the 1,2-trans glycosidic linkage. The o-hexenyl benzoate group activates the pentasaccharide donor 232 and is stable enough until it became activated with the gold(I) complex. The coupling reaction was performed under the catalysis of [Au(PPh3)(OTf)] in the presence of 5 Å molecular sieves in DCM at room temperature to afford the desired steroid pentasaccharide 234 in 80% yield (Scheme 45).
 | ||
| Scheme 45 | ||
The Nicolaou group reported187 a gold-catalyzed enantioselective total synthesis of the newly discovered antibiotics platensimycin and platencin. On the other hand, the chlorinated sesquiterpene, gomerones C 236 was isolated from the samples of Laurencia majuscula collected at the southern coast of La Gomera, Canary Islands and their structural elucidation was reported by a Spanish research group.188 The structurally interesting gomerane skeleton along with its unexplored biological activity made gomerone C a worthy target for synthesis.189 Recently, Carreira et al. reported a total synthesis of gomerone C by gold(1)-catalyzed cyclization as one of the key steps (Fig. 10).190
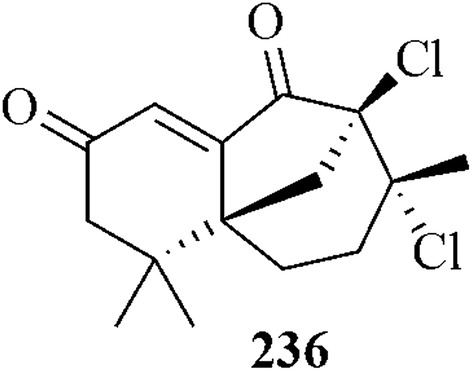 | ||
| Fig. 10 The chlorinated sesquiterpene, gomerones C | ||
Carreira et al. reported191 the first total synthesis of (−)-indoxamycin B (243), which is responsible for the structural reassignment of the natural product. This belongs to a novel class of polyketides and was first isolated from the saline cultures of marine-derived actinomycetes. Indoxamycin B displays growth inhibition against HT-29 tumor cell lines (IC50 = 0.59 mm and 0.31 mm, respectively).192 Its important biological activity along with a highly congested and stereochemically dense core render the indoxamycins as prime targets for synthetic studies. The indoxamycins possess a common tricyclic carbon skeleton. The tricyclic carbon skeleton of indoxamycin B bears six contiguous asymmetric centers, an α,β-unsaturated carboxylic acid side chain, and a trisubstituted alkene appendage. Three stereogenic centers are quaternary, and among these, two are vicinal.
Initially, their attempt to conduct the Saucy–Marbet rearrangement at elevated temperature (160 °C, o-xylene) failed to give the desired product. But the rearrangement proceeded smoothly by the use of the trinuclear Au(I)–oxo complex [(Ph3PAu)3O]BF4 as a catalyst (1 mol%). This reaction yielded allene 239 as the only detectable diastereomer (84%). Another intramolecular Au(I)-catalyzed hydroalkoxylation of allene 240 afforded the tetrahydrofuran ring of the indoxamycin framework. Thus the synthesis of tetracyclic intermediate 241 was accomplished in 72% yield and as a 3.2:1 mixture of inseparable diastereomers at C(2) (Scheme 46).
 | ||
| Scheme 46 | ||
5. Mixed coinage metal-catalyzed synthesis of natural products
Anisomycin shows antibiotic properties. This was isolated from the culture filtrates of two Streptomyces species e.g., S. griseolus and S. roseochromogenes.193 The natural product, (−)-anisomycin 247 blocks the peptide bond formation specifically in eukaryotic ribosomes. This has found use as a valuable tool in molecular biology.194 Additionally, it shows fungicidal activity as well as high in vitro antitumor activity.195 Maarseveen et al. used a combined copper-catalyzed Crabbe reaction and gold-catalyzed allene–amination reaction in the total synthesis of (−)-anisomycin. The Crabbe reaction of proparzylic amine 244 provided the allenic amine 245 in 60% yield. Cationic gold complex-catalyzed cyclization of 245 afforded 246 in 72% yield. It was found that on small scales the reaction is sensitive to traces of water and/or air oxygen. This is probably due to the hygroscopic nature of AgOTf. With the synthesis of 246 the formal synthesis of (+)-anisomycin was accomplished in only seven steps (Scheme 47).196 | ||
| Scheme 47 | ||
After successfully accomplishing the total synthesis of (−)-anisomycin the same group also attempted to synthesize another natural product (−)-Cytoxazone 248. This was isolated from the cultures of Streptomyces species and found to interfere with the production of cytokines IL4 and IL10 and IgG by selective inhibition of the signalling pathway in Th2 cells. They were able to efficiently synthesize the key 5-methyleneoxazolidin-2-one intermediate by gold-catalyzed cyclization of N-Boc-protected propargylic amines, but failed to successfully perform the hydroboration of the double bond to afford the (−)-cytoxazone.
Furanoid linalool oxide is used in perfumery and also for the reconstitution of essential oils.197 It is an important constituent of black and green tea and is also found in many essential oils198 and fruit aromas.199 On the other hand, isocyclocapitelline (253) and isochrysotricine (254), two other furanoid compounds, were isolated from the Rubiaceae plant Hedyotis capitellata; these are widely used in traditional Chinese and Vietnamese herb medicine.200 The biological activity studies of isochrysotricine and isocyclocapitelline were hampered by minute amounts of the alkaloids available from natural sources. However, chrysotricine is found to be active against the growth of HL-60 leukemia cells.201 An expedient synthesis of these natural products was achieved where the key steps are the copper-mediated SN2/-substitution of propargyl oxiranes 250 and the gold-catalyzed cycloisomerization of dihydroxyallenes 251, resulting in a highly efficient center-to-axis-to-center chirality transfer. The common key intermediate for the synthesis of isocyclocapitelline and isochrysotricine is 251. Initially, 251 was synthesized in excellent yield without any loss of stereochemical information by the treatment of epoxyalcohol ent-250 with Dess–Martin periodinane followed by Grignard addition and SN2′-substitution. Alternatively, the conversion of 250 to 251 can be carried out in a one-pot SN2′-substitution/1,2-addition by treating the substrate first with methylmagnesium cuprate and then with excess Grignard reagent. This sequence afforded the allenic diol 251 in 93% yield and with excellent stereocontrol (>98% ee). The subsequent axis-to-center chirality transfer was accomplished by gold-catalyzed cycloisomerization (0.05 mol% AuCl3 in THF) to give the key intermediate 252 in excellent yield (97%) and high stereochemical purity (98% ds, >98% ee) (Scheme 48).202
 | ||
| Scheme 48 | ||
6. Conclusion
This review article summarizes the collection of most recent advances in coinage metal-catalyzed (Cu, Ag and Au) reactions which were successfully utilized in the synthesis of natural products and their analogs. The methodologies applied often simplify the progress of the synthesis and the biological investigation of skeletally and structurally diverse natural products. High tolerance of functional groups along with relatively mild reaction conditions motivated the researchers to develop orthogonal catalytic systems using coinage metals for selective C–C, C–N, C–S or C–O bond formations. Moreover, the good σ- and/or π-Lewis acidity of both Ag(I) and Au(I) is used for easy substrate activation to produce important intermediates of different bioactive natural products. Sometimes, these reactions are very much effective in the construction of sterically congested chiral centers. We believe that this review has presented convincing justification for the need to develop more new coinage metal-catalyzed protocols and expand their applications in organic synthesis, especially in the synthesis of complex organic molecules and natural products.Acknowledgements
We are thankful to UGC, New Delhi (KCM for a UGC Emeritus Fellowship) and CSIR, New Delhi [BS for a Senior Research Fellowship (Ext.)].References
- (a) D. H. R. Barton, Chem. Rev., 1995, 95, 1675 CrossRef CAS; (b) K. Tatsuta and S. Hosokawa, Chem. Rev., 2005, 105, 4707 CrossRef CAS PubMed; (c) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed; (d) K. E. O. Ylijoki and J. M. Stryker, Chem. Rev., 2013, 113, 2244 CrossRef CAS PubMed; (e) B. B. Toure and D. G. Hall, Chem. Rev., 2009, 109, 4439 CrossRef CAS PubMed; (f) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395 CrossRef CAS PubMed; (g) Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar and S. K. Chattopadhyay, Wiley-VCH, 2011 Search PubMed.
- (a) K. Takao, R. Munakata and K. Tadano, Chem. Rev., 2005, 105, 4779 CrossRef CAS PubMed; (b) Z.-L. Song, C.-A. Fan and Y.-Q. Tu, Chem. Rev., 2011, 111, 7523 CrossRef CAS PubMed; (c) K. C. Majumdar and B. Sinha, Synthesis, 2013, 45, 1271 CrossRef CAS PubMed; (d) K. C. Majumdar and R. K. Nandi, Tetrahedron, 2013, 69, 6921 CrossRef CAS PubMed; (e) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563 CrossRef CAS PubMed; (f) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550 CrossRef CAS PubMed; (g) L. F. Silva Jr and B. Olofsson, Nat. Prod. Rep., 2011, 28, 1722 RSC; (h) E. Marques-Lopez, R. P. Herrera and M. Christmann, Nat. Prod. Rep., 2010, 27, 1138 RSC; (i) E. A. Ilardi, C. E. Stivala and A. Zakarian, Chem. Soc. Rev., 2009, 38, 3133 RSC; (j) K. C. Nicolaou, S. P. Ellery and J. S. Chen, Angew. Chem., Int. Ed., 2009, 48, 7140 CrossRef CAS PubMed.
- (a) B. H. Lipshutz and Y. Yamamoto, Chem. Rev., 2008, 108, 2793 CrossRef CAS PubMed; (b) R. A. Widenhoefer and X. Han, Eur. J. Org. Chem., 2006, 4555 CrossRef CAS; (c) A. Furstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef PubMed; (d) E. Jimenez-Nunez and A. M. Echavarren, Chem. Commun., 2007, 333 RSC; (e) N. Marion and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2750 CrossRef CAS PubMed; (f) J. Marco-Contelles and E. Soriano, Chem.–Eur. J., 2007, 13, 1350 CrossRef CAS PubMed; (g) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS PubMed.
- (a) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef CAS PubMed; (b) B. Alcaide, P. Almendros and J. M. Alonso, Molecules, 2011, 16, 7815–7843 CrossRef CAS PubMed.
- A. Shafir, P. A. Lichtor and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 3490 CrossRef CAS PubMed.
- (a) X. Xie, Y. Chen and D. Ma, J. Am. Chem. Soc., 2006, 128, 16050 CrossRef CAS PubMed; (b) H. Wehlan, M. Dauber, M.-T. Mujica Fernaud, J. Schuppan, S. Keiper, R. Mahrwald, B. Ziemer, M.-E. Juarez Garcia and U. Koert, Chem.–Eur. J., 2006, 12, 7378 CrossRef CAS PubMed.
- S. De Munari, P. Barassi, A. Cerri, G. Fedrizzi, M. Gobbini, M. Mabilia and P. Melloni, J. Med. Chem., 1998, 41, 3033 CrossRef CAS PubMed.
- (a) R. B. Turner, E. G. Herzog, R. B. Morin and A. Riebel, Tetrahedron Lett., 1959, 1, 7 CrossRef; (b) W. J. Gensler and G. M. Sherman, J. Am. Chem. Soc., 1959, 81, 5217 CrossRef CAS.
- S. Phoenix, M. S. Reddy and P. Deslongchamps, J. Am. Chem. Soc., 2008, 130, 13989 CrossRef CAS PubMed.
- (a) T. D. Sirakova, A. M. Titzmaurice and P. E. Kolattukudy, J. Bacteriol., 2002, 184, 6796 CrossRef CAS; (b) A. Rao and A. Ranganathan, Mol. Genet. Genomics, 2004, 272, 571 CrossRef CAS PubMed; (c) J. S. Cox, B. Chen, M. McNeil and W. R. Jacobs Jr, Nature, 1999, 402, 79 CrossRef CAS PubMed.
- E. Casas-Arce, B. Horst, B. L. Feringa and A. J. Minnaard, Chem.–Eur. J., 2008, 14, 4157 CrossRef CAS PubMed.
- (a) J. Su, Y. Zhong and L. Zeng, J. Nat. Prod., 1991, 54, 380 CrossRef CAS; (b) Z. Zeng and X. Xu, Tetrahedron Lett., 2000, 41, 3459 CrossRef CAS; (c) Q. Zhu, L. Qiao, Y. Wu and Y.-L. Wu, J. Org. Chem., 2001, 66, 2692 CrossRef CAS PubMed; (d) B. Sun and X. Xu, Tetrahedron Lett., 2005, 46, 8431 CrossRef CAS PubMed; (e) B. Sun and X. Xu, Tetrahedron Lett., 2006, 47, 299 CrossRef CAS PubMed.
- M. K. Brown and A. H. Hoveyda, J. Am. Chem. Soc., 2008, 130, 12904 CrossRef CAS PubMed.
- H. Ohkuma, N. Naruse, Y. Nishiyama, T. Tsuno, Y. Hoshino, Y. Sawada, M. Konishi and T. Oki, J. Antibiot., 1992, 45, 1239 CrossRef CAS.
- C. P. Burke, N. Haq and D. L. Boger, J. Am. Chem. Soc., 2010, 132, 2157 CrossRef CAS PubMed.
- S. Zheng, S. J. Aves, L. Laraia, W. R. J. Galloway, K. G. Pike, W. Wu and D. R. Spring, Chem.–Eur. J., 2012, 18, 3193 CrossRef CAS PubMed.
- (a) H. Y. Min, E. J. Park, J. Y. Hong, Y. J. Kang, S. J. Kim, H. J. Chung, E. R. Woo, T. M. Hung, U. J. Youn, Y. S. Kim, S. S. Kang, K. Bae and S. K. Lee, Bioorg. Med. Chem. Lett., 2008, 18, 523 CrossRef CAS PubMed; (b) L. Li, Q. Pan, M. Sun, Q. Lu and X. Hu, Life Sci., 2007, 80, 741 CrossRef CAS PubMed; (c) M. Huang, J. Jin, H. Sun and G. T. Liu, Cancer Chemother. Pharmacol., 2008, 62, 1015 CrossRef CAS PubMed.
- L. Opletal, H. Sovova and M. Bartlova, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2004, 812, 357 CAS.
- N. K. Kochetkov, A. Khorlin and O. S. Chizhov, Tetrahedron Lett., 1962, 3, 361 CrossRef.
- (a) D. F. Chen, S. X. Zhang, L. Xie, J. X. Xie, K. Chen, Y. Kashiwada, B. N. Zhou, P. Wang, L. M. Cosentino and K. H. Lee, Bioorg. Med. Chem., 1997, 5, 1715 CrossRef CAS; (b) W. H. Ma, Y. Lu, H. Huang, P. Zhou and D. F. Chen, Bioorg. Med. Chem. Lett., 2009, 19, 4958 CrossRef CAS PubMed.
- K. Y. Jung, I. S. Lee, S. R. Oh, D. S. Kim and H. K. Lee, Phytomedicine, 1997, 4, 229 CrossRef CAS.
- D. S. Surry, D. J. Fox, S. J. F. Macdonald and D. R. Spring, Chem. Commun., 2005, 2589 RSC.
- H. J. Ren, A. Krasovskiy and P. Knochel, Org. Lett., 2004, 6, 4215 CrossRef CAS PubMed.
- S. Djokic, G. Kobrehel, G. Lazarevski, N. Lopotar, Z. Tamburasev, B. Kamenar, A. Nagl and I. Vickovic, J. Chem. Soc., Perkin Trans. 1, 1986, 1881 RSC.
- R. B. Woodward, B. W. Au-Yeung, P. Balaram, L. J. Browne, D. E. Ward, B. W. Au-Yeung, P. Balaram, L. J. Browne, P. J. Card and C. H. Chen, J. Am. Chem. Soc., 1981, 103, 3215 CrossRef CAS.
- H. C. Kim and S. O. Kang, Angew. Chem., Int. Ed., 2009, 48, 1827 CrossRef CAS PubMed.
- (a) M. Isaka, N. Rugseree, P. Maithip, P. Kongsaeree, S. Prabpai and Y. Thebtaranonth, Tetrahedron, 2005, 61, 5577 CrossRef CAS PubMed; (b) M. Isaka, W. Prathumpai, P. Wongsa and M. Tanticharoen, Org. Lett., 2006, 8, 2815 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, D. Sarlah, T. R. Wu and W. Zhan, Angew. Chem., Int. Ed., 2009, 48, 6870 CrossRef CAS PubMed; (b) K. P. Reber, S. D. Tilley and E. J. Sorensen, Chem. Soc. Rev., 2009, 38, 3022 RSC; (c) M. Huang, L. Song and B. Liu, Org. Lett., 2010, 12, 2504 CrossRef CAS PubMed; (d) G. T. Halvorsen and W. R. Roush, Tetrahedron Lett., 2011, 52, 2072 CrossRef CAS PubMed; (e) K. C. Nicolaou, Y. Sun, D. Sarlah, W. Zhan and T. R. Wu, Org. Lett., 2011, 20, 5708 CrossRef PubMed.
- H. Uchiro, R. Kato, Y. Arai, M. Hasegawa and Y. Kobayakawa, Org. Lett., 2011, 13, 6268 CrossRef CAS PubMed.
- M. Wolter, G. Nordmann, G. E. Job and S. L. Buchwald, Org. Lett., 2002, 4, 973 CrossRef CAS PubMed.
- Y. Q. Ye, H. Koshino, J. Onose, K. Yoshikawa, N. Abe and S. Takahashi, Org. Lett., 2009, 11, 5074 CrossRef CAS PubMed.
- C. Xie, H. Koshino, Y. Esumi, J. Onose, K. Yoshikawa and N. Abe, Bioorg. Med. Chem. Lett., 2006, 16, 5424 CrossRef CAS PubMed.
- J. Pospsil, C. Muller and A. Furstner, Chem.–Eur. J., 2009, 15, 5956 CrossRef PubMed.
- (a) J.-P. Kinet, Annu. Rev. Immunol., 1999, 17, 931 CrossRef CAS PubMed; (b) P. Jardieu, Curr. Opin. Immunol., 1995, 7, 779 CrossRef CAS; (c) S. C. Garman, J.-P. Kinet and T. S. Jardetzky, Annu. Rev. Immunol., 1999, 17, 973 CrossRef CAS PubMed.
- (a) T. Fujita, K. Inoue, S. Yamamoto, T. Ikumoto, S. Sasaki, R. Toyama, K. Chiba, Y. Hoshino and T. Okumoto, J. Antibiot., 1994, 47, 216 CrossRef CAS; (b) S. Sasaki, R. Hashimoto, M. Kiuchi, K. Inoue, T. Ikumoto, R. Hirose, K. Chiba, Y. Hoshino, T. Okumoto and T. Fujita, J. Antibiot., 1994, 47, 420 CrossRef CAS; (c) M. Brunner and A. M. P. Koskinen, Curr. Org. Chem., 2004, 8, 1629 CrossRef CAS; (d) H.-S. Byun, X. Lu and R. Bittman, Synthesis, 2006, 2447 CAS.
- (a) D. Kluepfel, J. Bagli, H. Baker, M.-P. Charest, A. Kudelski, S. N. Sehgal and C. V. Zina, J. Antibiot., 1972, 25, 109 CrossRef CAS; (b) T. Fujita, K. Inoue, S. Yamamoto, T. Ikumoto, S. Sasaki, R. Toyama, K. Chiba, Y. Hoshino and T. Okumoto, J. Antibiot., 1994, 47, 208 CrossRef CAS.
- (a) T. Mashiko, K. Hara, D. Tanaka, Y. Fujiwara, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 11342 CrossRef CAS PubMed; (b) M. Shibasaki, M. Kanai, S. Matsunaga and N. Kumagai, Acc. Chem. Res., 2009, 42, 1117 CrossRef CAS PubMed; (c) A. Berkessel and H. Groger, Asymmetric Organocatalysis: from Biomimetic Concepts to Applications in Asymmetric Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed; (d) M. S. Taylor and E. N. Jacobsen, Angew. Chem., 2006, 118, 1550 (Angew. Chem., Int. Ed., 2006, 45, 1520) CrossRef.
- F. Berhal, S. Takechi, N. Kumagai and M. Shibasaki, Chem.–Eur. J., 2011, 17, 1915 CrossRef CAS PubMed.
- (a) D. C. Gournelif, G. G. Laskaris and R. Verpoorte, Nat. Prod. Rep., 1997, 14, 75 RSC; (b) M. M. Joullié and D. J. Richard, Chem. Commun., 2004, 2011 RSC; (c) N. H. Tan and J. Zhou, Chem. Rev., 2006, 106, 840 CrossRef CAS PubMed; (d) H. R. Hesham, R. El-Seedi, M. H. Zahra, U. Goransson and R. Verpoorte, Phytochem. Rev., 2007, 6, 143 CrossRef.
- (a) B. H. Ann and M. H. Park, Arch. Pharmacal Res., 1987, 10, 203 CrossRef; (b) B. H. Ann and M. H. Park, Arch. Pharmacal Res., 1987, 10, 208 CrossRef.
- (a) O. Blanpin, M. Pais and M. A. Quevauviller, Ann. Pharm. Fr., 1963, 21, 147 CAS; (b) A. F. Morel, C. A. Araujo, U. F. da Silva, S. C. S. M. Hoelzel, R. Záchia and N. R. Bastos, Phytochemistry, 2002, 61, 561 CrossRef CAS; (c) S. Suksamrarn, N. Suwannapoch, N. Aunchai, M. Kuno, P. Ratananukul, R. Haritakun, C. Jansakul and S. Ruchirawat, Tetrahedron, 2005, 61, 1175 CrossRef CAS PubMed.
- (a) J. C. Lagarias, R. A. Houghten and H. Rapoport, J. Am. Chem. Soc., 1978, 100, 8202 CrossRef CAS; (b) M. de Greef, S. Abeln, K. Belkasmi, A. Dömling, R. V. A. Orru and L. A. Wessjohann, Synlett, 2006, 3997 CAS.
- (a) S. P. East, F. Shao, L. Williams and M. M. Joullié, Tetrahedron, 1998, 54, 13371 CrossRef CAS; (b) B. H. Han, Y. C. Kim, M. K. Park, J. H. Park, H. J. Go, H. O. Yang, D. Y. Suh and Y. H. Kang, Heterocycles, 1995, 41, 1909 CrossRef CAS PubMed; (c) P. Cristau, T. Temal-Laïb, M. Bois-Choussy, M. T. Martin, J. Vors and J. P. Zhu, Chem.–Eur. J., 2005, 11, 2668 CrossRef CAS PubMed.
- M. Toumi, V. Rincheval, A. Young, D. Gergeres, E. Turos, F. Couty, B. Mignotte and G. Evano, Eur. J. Org. Chem., 2009, 3368 CrossRef CAS.
- (a) C. Gopalakrishnan, D. Shankaranarayan, L. Kameswaran and S. Natarajan, Indian J. Med. Res., 1979, 69, 513 CAS; (b) W. Gao, W. Lam, S. Zhong, C. Kaczmarek, D. C. Baker and Y.-C. Geng, Cancer Res., 2004, 64, 678 CrossRef CAS; (c) H.-S. Shiah, W. Gao, D. C. Baker and Y.-C. Cheng, Mol. Cancer Ther., 2006, 5, 2482 CrossRef PubMed; (d) Y. Fu, S. K. Lee, H.-Y. Min, T. Lee, J. Lee, M. Cheng and S. Kim, Bioorg. Med. Chem. Lett., 2007, 17, 97 CrossRef CAS PubMed.
- (a) E. S. Sherman, S. R. Chemler, T. B. Tan and O. Gerlits, Org. Lett., 2004, 6, 1573 CrossRef CAS PubMed; (b) E. S. Sherman, P. H. Fuller, D. Kasi and S. R. Chemler, J. Org. Chem., 2007, 72, 3896 CrossRef CAS PubMed; (c) W. Zeng and S. R. Chemler, J. Am. Chem. Soc., 2007, 129, 12948 CrossRef CAS PubMed; (d) P. H. Fuller and S. R. Chemler, Org. Lett., 2007, 9, 5477 CrossRef CAS PubMed.
- W. Zeng and S. R. Chemler, J. Org. Chem., 2008, 73, 6045 CrossRef CAS PubMed.
- (a) J. P. Karwowski, M. Jackson, R. J. Theriault, R. H. Chen, G. J. Barlow and M. L. Maus, J. Antibiot., 1989, 42, 506 CrossRef CAS; (b) R. M. Fronko, J. C. Lee, J. G. Galazzo, S. Chamberland, F. Malouin and M. D. Lee, J. Antibiot., 2000, 53, 1405–1410 CrossRef CAS.
- (a) F. Isono and M. Inukai, Antimicrob. Agents Chemother., 1991, 35, 234 CrossRef CAS; (b) M. Inukai, F. Isono and A. Takatsuki, Antimicrob. Agents Chemother., 1993, 37, 980 CrossRef CAS.
- (a) T. D. H. Bugg, A. J. Lloyd and D. I. Roper, Infect. Disord.: Drug Targets, 2006, 6, 85 CrossRef CAS; (b) A. Bouhss, A. E. Trunkfield, T. D. Bugg and D. Mengin-Lecreulx, FEMS Microbiol. Rev., 2008, 32, 208 CrossRef CAS PubMed; (c) B. Al-Dabbagh, X. Henry, M. El Ghachi, G. Auger, D. Blanot, C. Parquet, D. Mengin-Lecreulx and A. Bouhss, Biochemistry, 2008, 47, 8919 CrossRef CAS PubMed.
- (a) M. Winn, R. J. M. Goss, K.-I. Kimura and T. D. H. Bugg, Nat. Prod. Rep., 2010, 27, 279 RSC; (b) K.-I. Kimura and T. D. H. Bugg, Nat. Prod. Rep., 2003, 20, 252 RSC.
- G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef CAS PubMed.
- A. Klapars, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 7421 CrossRef CAS PubMed.
- K. Okamoto, M. Sakagami, F. Feng, H. Togame, H. Takemoto, S. Ichikawa and A. Matsuda, Org. Lett., 2011, 13, 5240 CrossRef CAS PubMed.
- A. R. Pereira, Z. Cao, N. Engene, I. E. Soria-Mercado, T. F. Murray and W. H. Gerwick, Org. Lett., 2010, 12, 4490 CrossRef CAS PubMed.
- N. Matsumori, D. Kaneno, M. Murata, H. Nakamura and K. Tachibana, J. Org. Chem., 1999, 64, 866 CrossRef CAS PubMed.
- R. Tello-Aburto, E. M. Johnson, C. K. Valdez and W. A. Maio, Org. Lett., 2012, 14, 2150 CrossRef CAS PubMed.
- L. Jiang, G. E. Job, A. Klapars and S. L. Buchwald, Org. Lett., 2003, 5, 3667 CrossRef CAS PubMed.
- (a) H. Luesch, W. Y. Yoshida, R. E. Moore, V. J. Paul and T. H. Corbett, J. Am. Chem. Soc., 2001, 123, 5418 CrossRef CAS PubMed; (b) D. Klein, J. C. Braekman, D. Daloze, L. Hoffmann, G. Castillo and V. Demoulin, J. Nat. Prod., 1999, 62, 934 CrossRef CAS PubMed; (c) S. Matthew, L. A. Salvador, P. J. Schupp, V. J. Paul and H. Luesch, J. Nat. Prod., 2010, 73, 1544 CrossRef CAS PubMed.
- M. Toumi, F. Couty, J. Marrot and G. Evano, Org. Lett., 2008, 10, 5027 CrossRef CAS PubMed.
- R. H. Jiao, S. Xu, J. Y. Liu, H. M. Ge, H. Ding, C. Xu, H. L. Zhu and R. X. Tan, Org. Lett., 2006, 8, 5709 CrossRef CAS PubMed.
- (a) B. K. S. Yeung, Y. Nakao, R. B. Kinnel, J. R. Carney, W. Y. Yoshida, P. J. Scheuer and M. Kelly-Borges, J. Org. Chem., 1996, 61, 7168 CrossRef CAS PubMed; (b) Y. Nakao, J. Kuo, W. Y. Yoshida, M. Kelly and P. J. Scheuer, Org. Lett., 2003, 5, 1387 CrossRef CAS PubMed.
- (a) A. Klapars, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 7421 CrossRef CAS PubMed; (b) A. Coste, M. Toumi, K. Wright, V. Razafimahaleó, F. Couty, J. Marrot and G. Evano, Org. Lett., 2008, 10, 3841 CrossRef CAS PubMed.
- (a) J. P. Wolfe, R. A. Rennels and S. L. Buchwald, Tetrahedron, 1996, 52, 7525 CrossRef CAS; (b) A. Correa, S. Elmore and C. Bolm, Chem.–Eur. J., 2008, 14, 3527 CrossRef CAS PubMed.
- (a) L. Rahbaek, J. Breinholt, J. C. Frisvad and C. Christophersen, J. Org. Chem., 1999, 64, 1689 CrossRef; (b) J.-R. Dai, B. K. Carte, P. J. Sidebottom, A. L. S. Yew, S.-B. Ng, Y. Huang and M. S. Butler, J. Nat. Prod., 2001, 64, 125 CrossRef CAS PubMed; (c) D. Zhang, X. Yang, J. S. Kang, H. D. Choi and B. W. Son, J. Antibiot., 2008, 61, 40 CrossRef CAS PubMed.
- R. Ookura, K. Kito, T. Ooi, M. Namikishi and T. Kusumi, J. Org. Chem., 2008, 73, 4245 CrossRef CAS PubMed.
- D. S. Bose and M. V. Chary, Synthesis, 2010, 643 CrossRef CAS PubMed.
- (a) X. Liu, H. Fu, Y. Jiang and Y. Zhao, Angew. Chem., Int. Ed., 2009, 48, 348 CrossRef CAS PubMed; (b) A. D. Roy, A. Subramanian and R. Roy, J. Org. Chem., 2006, 71, 382 CrossRef CAS PubMed; (c) A. H. Amin, D. R. Mehta and S. S. Samarth, Prog. Drug. Res., 1970, 14, 218 Search PubMed; (d) S. Sinha and M. Srivastava, Prog. Drug. Res., 1994, 43, 143 CAS.
- U. A. Kshirsagar and N. P. Argade, Org. Lett., 2010, 12, 3716 CrossRef CAS PubMed.
- P. E. Zhichkin, X. Jin, H. Zhang, L. H. Peterson, C. Ramirez, T. M. Snyder and H. S. Burton, Org. Biomol. Chem., 2010, 8, 1287 CAS.
- (a) M. R. Boyd, C. Farina, P. Belfiore, S. Gagliardi, J. W. Kim, Y. Hayakawa, J. A. Beutler, T. C. McKee, B. J. Bowman and E. J. Bowman, J. Pharmacol. Exp. Ther., 2001, 297, 114 CAS; (b) L. Yet, Chem. Rev., 2003, 103, 4283 CrossRef CAS PubMed; (c) J. W. Kim, K. Shin-ya, K. Furihata, Y. Hayakawa and H. Seto, J. Org. Chem., 1999, 64, 153 CrossRef CAS PubMed.
- M. Forgac, Nat. Rev. Mol. Cell Biol., 2007, 8, 917 CrossRef CAS PubMed.
- X. Wang and J. A. Porco, J. Am. Chem. Soc., 2003, 125, 6040 CrossRef CAS PubMed.
- G. A. Molander and F. Dehmel, J. Am. Chem. Soc., 2004, 126, 10313 CrossRef CAS PubMed.
- C. M. Schneider, K. Khownium, W. Li, J. T. Spletstoser, T. Haack and G. I. Georg, Angew. Chem., Int. Ed., 2011, 50, 7855 CrossRef CAS PubMed.
- D. M. Brestensky, D. E. Huseland, C. McGettigan and J. M. Stryker, Tetrahedron Lett., 1988, 29, 3749 CrossRef CAS.
- (a) J. Kabbara, C. Hoffmann and D. Schinzer, Synthesis, 1995, 299 CrossRef CAS PubMed; (b) F. von der Ohe and R. Bruckner, New J. Chem., 2000, 24, 659 RSC; (c) D. S. Rawat and J. M. Zaleski, Synth. Commun., 2002, 32, 1489 CrossRef CAS PubMed.
- M. Brichacek, L. A. Batory, N. A. McGrath and J. T. Njardarson, Tetrahedron, 2010, 66, 4832–4840 CrossRef CAS PubMed.
- (a) S.-G. Cao, X.-H. Wu, K.-Y. Sim, B. K. H. Tan, J. T. Pereira and S.-H. Goh, Tetrahedron, 1998, 54, 2143 CrossRef CAS; (b) H. Yoda, Y. Nakaseko and K. Takabe, Synlett, 2002, 1532 CrossRef CAS PubMed; (c) M. C. Carreno, G. Hernandez-Torres, A. Urbano and F. Colobert, Org. Lett., 2005, 7, 5517 CrossRef CAS PubMed; (d) K. R. Prasad and S. L. Gholap, J. Org. Chem., 2006, 71, 3643 CrossRef CAS PubMed; (e) S. Ghosh, C. N. Rao and S. K. Dutta, Synlett, 2007, 1464 CrossRef CAS PubMed.
- M. Brichacek, L. A. Batory and J. T. Njardarson, Angew. Chem., Int. Ed., 2010, 49, 1648 CrossRef CAS PubMed.
- M. R. Truter, D. E. Fenton and B. L. Vickery, J. Chem. Soc. D, 1971, 93 Search PubMed.
- A. Ortega, J. F. Blount and P. S. Manchand, J. Chem. Soc., Perkin Trans. 1, 1982, 10, 2505 RSC.
- A. Padwa, D. J. Austin, S. F. Hornbuckle, M. A. Semones, M. P. Doyle and M. N. Protopopova, J. Am. Chem. Soc., 1992, 114, 1874 CrossRef CAS.
- S. Levin, R. R. Nani and S. E. Reisman, Org. Lett., 2010, 12, 780 CrossRef CAS PubMed.
- S. G. Liao, Z. J. Zhan, S. P. Yang and J. M. Yue, Org. Lett., 2005, 7, 1379 CrossRef CAS PubMed.
- (a) C. M. Hasler, G. Acs and P. Blumberg, Cancer Res., 1992, 52, 202 CAS; (b) Z. Q. Lu, S. H. Guan, X. N. Li, G. T. Chen, J. Q. Zhang, H. L. Huang, X. Liu and D. A. Guo, J. Nat. Prod., 2008, 71, 873 CrossRef CAS PubMed; (c) M. M. Ana, G. Nora, R. A. Jose, M. A. Pedro, M. Joseph and U. F. Maria-Jose, J. Nat. Prod., 2006, 69, 950 CrossRef PubMed.
- L. F. Yu, H. N. Hu and F. J. Nan, J. Org. Chem., 2011, 76, 1448 CrossRef CAS PubMed.
- (a) C. Zhang, J. Ondeyka, Z. Guan, L. Dietrich, B. Burgess, J. Wang and S. B. Singh, J. Antibiot., 2009, 62, 699 CrossRef CAS PubMed; (b) K. B. Herath, C. Zhang, H. Jayasuriya, J. G. Ondeyka, D. L. Zink, B. Burgess, J. Wang and S. B. Singh, Org. Lett., 2008, 10, 1699 CrossRef CAS PubMed; (c) Z. Yu, M. J. Smanski, R. M. Peterson, K. Marchillo, D. Andes, S. R. Rajski and B. Shen, Org. Lett., 2010, 12, 1744 CrossRef CAS PubMed; (d) C. Zhang, J. Ondeyka, L. Dietrich, F. P. Gailliot, M. Hesse, M. Lester, K. Dorso, M. Motyl, S. N. Ha, J. Wang and S. B. Singh, Bioorg. Med. Chem., 2010, 18, 2602 CrossRef CAS PubMed.
- N. A. McGrath, E. S. Bartlett, S. Sittihan and J. T. Njardarson, Angew. Chem., Int. Ed., 2009, 48, 8543 CrossRef CAS PubMed.
- S. Hirai and M. Nakada, Tetrahedron, 2011, 67, 518 CrossRef CAS PubMed.
- (a) U. Rinner and T. Hudlicky, Synlett, 2005, 365 CAS; (b) Z. Jin, Nat. Prod. Rep., 2005, 22, 111 RSC; (c) Y. Chapleur, F. Chretien, S. Ibn Ahmed and M. Khaldi, Curr. Org. Synth., 2006, 3, 341 CrossRef CAS.
- (a) G. R. Pettit and N. Melody, J. Nat. Prod., 2005, 68, 207 CrossRef CAS PubMed; (b) G. Pettit, S. A. Eastham, N. Melody, B. Orr, D. L. Herald, J. McGregor, J. C. Knight, D. L. Doubek, G. R. Pettit III, L. C. Garner and J. A. Bell, J. Nat. Prod., 2006, 69, 7 CrossRef CAS PubMed.
- C. K. Jana and A. Studer, Chem.–Eur. J., 2008, 14, 6326 CrossRef CAS PubMed.
- K. Kong, Z. Moussa, C. Lee and D. Romo, J. Am. Chem. Soc., 2011, 133, 19844 CrossRef CAS PubMed.
- R. Munday, N. R. Towers, L. Mackenzie, V. Beuzenberg, P. T. Holland and C. O. Miles, Toxicon, 2004, 44, 173 CrossRef CAS PubMed.
- R. Araoz, D. Servent, J. Molgo, B. I. Iorga, C. Fruchart-Gaillard, E. Benoit, Z. Gu, C. Stivala and A. J. Zakarian, J. Am. Chem. Soc., 2011, 133, 10499 CrossRef CAS PubMed.
- J. Jaupovic and F. Eid, Phytochemistry, 1987, 26, 2427 CrossRef.
- R. N. Daniels, O. O. Fadeyi and C. W. Lindsley, Org. Lett., 2008, 10, 4097 CrossRef CAS PubMed.
- O. Olugbeminiyi, R. Fadeyi, N. Daniels, S. M. DeGuire and C. W. Lindsley, Tetrahedron Lett., 2009, 50, 3084 CrossRef PubMed.
- T. Shinada, K. Oe and Y. Ohfune, Tetrahedron Lett., 2012, 53, 3250 CrossRef CAS PubMed.
- (a) J. Kobayashi, F. Kanda, M. Ishibashi and H. Shigemori, J. Org. Chem., 1991, 56, 4574 CrossRef CAS; (b) T. Jahn, G. M. Konig, A. D. Wright, G. Worheide and J. Reitner, Tetrahedron Lett., 1997, 38, 3883 CrossRef CAS.
- (a) W. Oppolzer, Tetrahedron, 1987, 43, 1969 CrossRef CAS; (b) W. Oppolzer, Pure Appl. Chem., 1990, 62, 1241 CrossRef CAS; (c) H. Ü. Kaniskan and P. Garner, J. Am. Chem. Soc., 2007, 129, 15460 CrossRef CAS PubMed.
- A. R. Rodriguez and B. W. Spur, Tetrahedron Lett., 2012, 53, 86 CrossRef CAS PubMed.
- (a) M. Zmijewski Jr and M. Goebel, J. Antibiot., 1982, 35, 524 CrossRef CAS; (b) S. Cang, S. Ohta, H. Chiba, O. Johda, H. Nomura, Y. Nagamatsu and A. Yoshimoto, J. Antibiot., 2001, 54, 304 CrossRef CAS; (c) J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669 CrossRef CAS PubMed.
- P. Siengalewicz, U. Rinne and J. Mulzer, Chem. Soc. Rev., 2008, 37, 2676 RSC.
- (a) D. A. Evans, C. R. Illig and J. C. Saddler, J. Am. Chem. Soc., 1986, 108, 2478 CrossRef CAS PubMed; (b) T. Fukuyama, L. Li, A. A. Laird and K. Frank, J. Am. Chem. Soc., 1987, 109, 1587 CrossRef CAS.
- P. Garner, H. U. Kaniskan, C. M. Keyari and L. Weerasinghe, J. Org. Chem., 2011, 76, 5283 CrossRef CAS PubMed.
- (a) O. Hoshino, The Alkaloids, ed. G. A. Cordell, Academic Press, New York, 1998, vol. 51, p. 323 Search PubMed; (b) S. F. Martin, The Alkaloids, ed. A. Brossi, Academic Press, New York, 1987, vol. 30, p. 251 Search PubMed.
- N. T. N. Tram, T. V. Titorenkova, V. S. Bankova, N. V. Handjieva and S. S. Popov, Fitoterapia, 2002, 73, 183 CrossRef.
- (a) R. V. K. Rao and J. V. L. N. Sheshagiri Rao, Curr. Sci., 1979, 48, 110 CAS; (b) P. Pacheco, M. Silva, W. Steglich and W. H. Watson, Rev. Latinoam. Quim., 1978, 9, 28 CAS; (c) S. M. Toaima, Alexandria J. Pharm. Sci., 2007, 21, 61 Search PubMed.
- G. Pandey, N. R. Gupta and S. R. Gadre, Eur. J. Org. Chem., 2011, 740 CrossRef CAS.
- H. Cong and J. A. Porco Jr, Org. Lett., 2012, 14, 2516 CrossRef CAS PubMed.
- H. Cong, C. F. Becker, S. J. Elliott, M. W. Grinstaff and J. A. Porco, J. Am. Chem. Soc., 2010, 132, 7514 CrossRef CAS PubMed.
- C. F. Chee, Y. K. Lee, M. J. C. Buckle and N. A. Rahman, Tetrahedron Lett., 2011, 52, 1797 CrossRef CAS PubMed.
- Y. Hano, J. Yamanaka, T. Nomura and Y. Momose, Heterocycles, 1995, 41, 1035 CrossRef CAS PubMed.
- S. G. Sudrik, N. K. Chaki, V. B. Chavan, S. P. Chavan, S. P. Chavan, H. R. Sonawane and K. Vijayamohanan, Chem.–Eur. J., 2006, 12, 859 CrossRef CAS PubMed.
- K. Mallick, M. Witcomb and M. Scurrell, Mater. Chem. Phys., 2006, 97, 283 CrossRef CAS PubMed.
- A. A. Sierle, D. B. Sierle and K. Kelly, J. Org. Chem., 2006, 71, 5357 CrossRef PubMed.
- (a) X. Wu, J. Zhou and B. B. Snider, Angew. Chem., 2009, 121, 1309 (Angew. Chem., Int. Ed., 2009, 48, 1283) CrossRef; (b) T. N. Snaddon, P. Buchgraber, S. Schulthoff, C. Wirtz, R. Mynott and A. Furstner, Chem.–Eur. J., 2010, 16, 12133 CrossRef CAS PubMed; (c) N. Stiasni and M. Hiersemann, Synlett, 2009, 2133 CAS; (d) Z. E. Wilson and M. A. Brimble, Org. Biomol. Chem., 2010, 8, 1284 RSC; (e) T. A. Wenderski, M. A. Marsini and T. R. R. Pettus, Org. Lett., 2011, 13, 118 CrossRef CAS PubMed.
- X. Wu, J. Zhou and B. B. Snider, J. Org. Chem., 2009, 74, 6245 CrossRef CAS PubMed.
- C. F. Bender, F. K. Yoshimoto, C. L. Paradise and J. K. de Brabander, J. Am. Chem. Soc., 2009, 131, 11350 CrossRef CAS PubMed.
- Y. Yamamoto, J. Org. Chem., 2007, 72, 7817 CrossRef CAS PubMed.
- F. J. Fananas, A. Mendoza, T. Arto, B. Temelli and F. Rodriguez, Angew. Chem., Int. Ed., 2012, 51, 4930 CrossRef CAS PubMed.
- J. Barluenga, A. Mendoza, F. Rodriguez and F. J. Fananas, Angew. Chem., 2009, 121, 1672 (Angew. Chem., Int. Ed., 2009, 48, 1644) CrossRef.
- (a) J. Kobayashi and M. Tsuda, Nat. Prod. Rep., 2004, 21, 77 RSC; (b) J. Kobayashi, J. Antibiot., 2008, 61, 271 CrossRef CAS PubMed.
- (a) T. Kubota, M. Tsuda and J. Kobayashi, Org. Lett., 2001, 3, 1363 CrossRef CAS PubMed; (b) T. Kubota, Y. Sakuma, M. Tsuda and J. Kobayashi, Mar. Drugs, 2004, 2, 83 CrossRef CAS; (c) T. Kubota, A. Suzuki, M. Yamada, S. Baba and J. Kobayashi, Heterocycles, 2010, 82, 333 CrossRef CAS.
- S. Mahapatra and R. G. Carter, Angew. Chem., Int. Ed., 2012, 51, 7948 CrossRef CAS PubMed.
- A. Buzas, F. Istrate and F. Gagosz, Org. Lett., 2006, 8, 1957 CrossRef CAS PubMed.
- F. Volz and N. Krause, Org. Biomol. Chem., 2007, 5, 1519 CAS.
- H. Morita, S. Oshimi, Y. Hirasawa, K. Koysma, T. Honda, W. Ekasari, G. Indrayanto and N. C. Zaini, Org. Lett., 2007, 9, 3691 CrossRef CAS PubMed.
- Y. S. Yao and Z. J. Yao, J. Org. Chem., 2008, 73, 5221 CrossRef CAS PubMed.
- (a) F. Baquero and F. Moreno, FEMS Microbiol. Lett., 1984, 23, 117 CrossRef CAS; (b) R. W. Jack and G. Jung, Curr. Opin. Chem. Biol., 2000, 4, 310 CrossRef CAS; (c) S. Duquesne, D. Destoumieux-Garzon, J. Peduzzi and S. Rebuffat, Nat. Prod. Rep., 2007, 24, 708 RSC.
- (a) W. M. Parks, A. R. Bottrill, O. A. Pierrat, M. C. Durrant and A. Maxwell, Biochimie, 2007, 89, 500 CrossRef CAS PubMed; (b) O. A. Pierrat and A. Maxwell, Biochemistry, 2005, 44, 4204 CrossRef CAS PubMed; (c) O. A. Pierrat and A. Maxwell, J. Biol. Chem., 2003, 278, 35016 CrossRef CAS PubMed.
- (a) A. Bayer, S. Freund, G. Nicholson and G. Jung, Angew. Chem., Int. Ed. Engl., 1993, 32, 1336 CrossRef; (b) P. Yorgey, J. Lee, J. Kordel, E. Vivas, P. Warner, D. Jebaratnam and R. Kolter, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 4519 CrossRef CAS.
- (a) S. Aimoto, Biopolymers, 1999, 51, 247 CrossRef CAS; (b) G. Chen, Q. Wan, Z. P. Tan, C. Kan, Z. H. Hua, K. Ranganathan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 7383 CrossRef CAS PubMed; (c) R. J. Payne, S. Ficht, W. A. Greenberg and C.-H. Wong, Angew. Chem., Int. Ed., 2008, 47, 4411 CrossRef CAS PubMed.
- M. Inoue, N. Shinohara, S. Tanabe, T. Takahashi, K. Okura, H. Itoh, Y. Mizoguchi, M. Iida, N. Lee and S. Matsuoka, Nat. Chem., 2010, 2, 280 CrossRef CAS PubMed.
- R. E. Thompson, K. A. Jolliffe and R. J. Payne, Org. Lett., 2011, 13, 680 CrossRef CAS PubMed.
- (a) C. V. Galliford and K. A. Scheidt, J. Org. Chem., 2007, 72, 1811 CrossRef CAS PubMed; (b) F. Ragaini, S. Cenini, D. Brignoli, M. Gasperini and E. Gallo, J. Org. Chem., 2003, 68, 460 CrossRef CAS PubMed.
- (a) G. W. Chan, T. Francis, D. R. Thureen, P. H. Offen, N. J. Pierce, J. W. Westley, R. K. Johnson and D. J. Faulkner, J. Org. Chem., 1993, 58, 2544 CrossRef CAS; (b) T. A. Berkhout, L. M. Havekes, N. J. Pearce and P. H. E. Groot, Biochem. J., 1990, 272, 181 CAS.
- (a) H. Fan, J. Peng, M. T. Hamann and J.-F. Hu, Chem. Rev., 2008, 108, 264 CrossRef CAS PubMed; (b) M. G. Banwell, T. E. Goodwin, S. Ng, J. A. Smith and D. J. Wong, Eur. J. Org. Chem., 2006, 3043 CrossRef CAS; (c) A. Furstner, M. M. Domostoj and B. Scheiper, J. Am. Chem. Soc., 2006, 128, 8087 CrossRef PubMed.
- Q. Li, A. Fan, Z. Lu, Y. Cui, W. Lin and Y. Jia, Org. Lett., 2010, 12, 4066 CrossRef CAS PubMed.
- (a) F. Tomita, K. Takahashi and K. Shimizu, J. Antibiot., 1983, 463 CrossRef CAS; (b) K. Takahashi and F. Tomita, J. Antibiot., 1983, 468 CrossRef CAS; (c) F. Tomita, K. Takahashi and T. Tamaoki, J. Antibiot., 1984, 1268 CrossRef CAS.
- T. Fukuyama and J. J. Nunes, J. Am. Chem. Soc., 1988, 110, 5196 CrossRef CAS.
- (a) P. Garner, W. B. Ho and H. Shin, J. Am. Chem. Soc., 1992, 114, 2767 CrossRef CAS; (b) P. Garner, W. B. Ho and H. Shin, J. Am. Chem. Soc., 1993, 115, 10742 CrossRef CAS; (c) S. Kwon and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 16796 CrossRef CAS PubMed.
- Y. C. Wu, M. Liron and J. Zhu, J. Am. Chem. Soc., 2008, 130, 7148 CrossRef CAS PubMed.
- (a) J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734 RSC; (b) C. D. Johnson, Acc. Chem. Res., 1993, 26, 476 CrossRef CAS.
- (a) K. T. Mead and B. N. Brewer, Curr. Org. Chem., 2003, 7, 227 CrossRef CAS; (b) J. E. Aho, P. M. Pihko and T. K. Rissa, Chem. Rev., 2005, 105, 4406 CrossRef CAS PubMed; (c) Y. Booth, W. Kitching and J. De Voss, Nat. Prod. Rep., 2009, 26, 490 RSC.
- G. Zinzalla, L.-G. Milroy and S. V. Ley, Org. Biomol. Chem., 2006, 4, 1977 CAS.
- X. Li, Y. Yao, Y. Zheng, I. Sattler and W. Lin, Arch. Pharmacal Res., 2007, 30, 812 CrossRef CAS.
- S. Chang and R. Britton, Org. Lett., 2012, 14, 5844 CrossRef CAS PubMed.
- B. Kang, J. Mowat, T. Pinter and R. Britton, Org. Lett., 2009, 11, 1717 CrossRef CAS PubMed.
- (a) E. J. Schantz and N. Y. Ann, Ann. N. Y. Acad. Sci., 1986, 479, 15 CrossRef CAS; (b) J. B. Tucker, International Security, 2002, 27, 107 CrossRef.
- R. B. Woodward and F. V. Brutcher Jr, J. Am. Chem. Soc., 1958, 80, 209 CrossRef CAS.
- S. H. Kang and D. H. Ryu, Tetrahedron Lett., 1997, 38, 607 CrossRef CAS.
- V. R. Bhonde and R. E. Looper, J. Am. Chem. Soc., 2011, 133, 20172 CrossRef CAS PubMed.
- H. Waldmann, L. Eberhardt, K. Wittsteina and K. Kumar, Chem. Commun., 2010, 46, 4622 RSC.
- Y. Wang, C. Kong, Y. Du, H. Song, D. Zhang and Y. Qin, Org. Biomol. Chem., 2012, 10, 2793 CAS.
- (a) T.-C. Chou, K. M. Depew, Y.-H. Zheng, M. L. Safer, D. Chan, B. Helfrich, D. Zatorska, A. Zatorski, W. Bornmann and S. J. Danishefsky, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8369 CrossRef CAS; (b) J. P. Karwowski, M. Jackson, R. R. Rasmussen, P. E. Humphrey, J. B. Poddig, W. L. Kohl, M. H. Scherr, S. Kadam and J. B. McAlpine, J. Antibiot., 1993, 46, 374 CrossRef CAS.
- (a) Modern Carbonyl Chemistry, ed. J. Otera, Wiley-VCH, Weinheim, 2000 Search PubMed; (b) L. Hintermann and A. Labonne, Synthesis, 2007, 1121 CrossRef CAS PubMed; (c) M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2011, 47, 6536 RSC; (d) N. D. Shapiro and F. D. Toste, Synlett, 2010, 5, 675 Search PubMed; (e) F. Gagosz, Tetrahedron, 2009, 65, 1757 CrossRef CAS PubMed; (f) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef PubMed.
- (a) A. S. K. Hashmi and M. Rudolph, Chem. Soc. Rev., 2008, 37, 1766 RSC; (b) A. Fürstner, Chem. Soc. Rev., 2009, 28, 3208 RSC; (c) B. Alcaide, P. Almendros and J. M. Alonso, Org. Biomol. Chem., 2011, 9, 4405 RSC.
- (a) B. Liu and J. K. De Brabander, Org. Lett., 2006, 8, 4907 CrossRef CAS PubMed; (b) Y. Li, F. Zhou and C. J. Forsyth, Angew. Chem., 2007, 119, 283 (Angew. Chem., Int. Ed., 2007, 46, 279) CrossRef; (c) H. H. Jung and P. E. Floreancig, J. Org. Chem., 2007, 72, 7359 CrossRef CAS PubMed; (d) B. M. Trost and G. Dong, Nature, 2008, 456, 485 CrossRef CAS PubMed; (e) B. M. Trost, B. M. O'Doyle and D. Hund, J. Am. Chem. Soc., 2009, 131, 15061 CrossRef CAS PubMed; (f) K. C. Fortner, D. Kato, Y. Tanaka and M. D. Shair, J. Am. Chem. Soc., 2010, 132, 275 CrossRef CAS PubMed; (g) C. Fang, Y. Pang and C. J. Forsyth, Org. Lett., 2010, 12, 4528 CrossRef CAS PubMed; (h) S. F. Tlais and G. B. Dudley, Org. Lett., 2010, 12, 4698 CrossRef CAS PubMed.
- E. P. Stout, A. P. Hasemeyer, A. L. Lane, T. M. Daenport, S. Engel, M. E. Hay, C. R. Fairchild, J. Prudhomme, K. L. Roch, W. Aalbersberg and J. Kubanek, Org. Lett., 2009, 11, 225 CrossRef CAS PubMed.
- (a) A. Leyva and A. Corma, J. Org. Chem., 2009, 74, 2067 CrossRef CAS PubMed; (b) C. Nieto-Oberhuber, S. Lopez and A. M. Echavarren, J. Am. Chem. Soc., 2005, 127, 6178 CrossRef CAS PubMed; (c) N. Mezailles, L. Ricard and F. Gagosz, Org. Lett., 2005, 7, 4133 CrossRef CAS PubMed.
- (a) W. E. Brenzovich Jr, Angew. Chem., Int. Ed., 2012, 51, 8933 CrossRef PubMed; (b) W. Chaładaj, M. Corbet and A. Furstner, Angew. Chem., Int. Ed., 2012, 51, 6929 CrossRef PubMed.
- (a) B. Bister, D. Bischoff, M. Strçbele, J. Riedlinger, A. Reicke, F. Wolter, A. T. Bull, H. P. Fiedler and R. D. Sussmuth, Angew. Chem., 2004, 116, 2628 (Angew. Chem., Int. Ed., 2004, 43, 2574) CrossRef; (b) K. C. Nicolaou, J. S. Chen, D. J. Edmonds and A. A. Estrada, Angew. Chem., 2009, 121, 670 (Angew. Chem., Int. Ed., 2009, 48, 660) CrossRef.
- (a) J. Riedlinger, A. Riecke, H. Zahner, B. Krismer, A. T. Bull, L. A. Maldonado, A. C. Ward, M. Goodfellow, B. Bister, D. Bischoff, R. D. Sussmuth and H.-P. Fiedler, J. Antibiot., 2004, 57, 271 CrossRef CAS; (b) S. Keller, H. S. Schadt, I. Ortel and R. D. Sussmuth, Angew. Chem., 2007, 119, 8433 (Angew. Chem., Int. Ed., 2007, 46, 8284) CrossRef.
- (a) K. C. Nicolaou and J. S. Chen, Classics in Total Synthesis III, Wiley-VCH, Veinheim, 2011 Search PubMed; (b) C. W. Zapf, B. A. Harrison, C. Drahl and E. J. Sorensen, Angew. Chem., 2005, 117, 6691 ( Angew. Chem., Int. Ed., 2005, 44, 6533) CrossRef; (c) K. C. Nicolaou and S. T. Harrison, J. Am. Chem. Soc., 2007, 129, 429 CrossRef CAS PubMed; (d) K. C. Nicolaou, S. T. Harrison and J. S. Chen, Synthesis, 2009, 33 CrossRef CAS PubMed; (e) E. A. Couladouros, E. A. Bouzas and A. D. Magos, Tetrahedron, 2006, 62, 5272 CrossRef CAS PubMed; (f) B. B. Snider and Y. Zhou, Org. Lett., 2005, 7, 4939 CrossRef CAS PubMed; (g) J.-P. Rath, M. Eipert, S. Kinast and M. E. Maier, Synlett, 2005, 314 CAS; (h) A. L. Zografos, A. Yiotakis and D. Georgiadis, Org. Lett., 2005, 7, 4515 CrossRef CAS PubMed.
- F. Bihelovic and R. N. Saicic, Angew. Chem., Int. Ed., 2012, 51, 5687 CrossRef CAS PubMed.
- (a) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395 CrossRef CAS PubMed; (b) A. S. K. Hashmi and M. Rudolph, Chem. Soc. Rev., 2008, 37, 1766 RSC; (c) N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776 RSC; (d) M. Rudolph and S. Hashmi, Chem. Soc. Rev., 2012, 41, 2448 RSC.
- N. Mezailles, L. Ricard and F. Gagosz, Org. Lett., 2005, 7, 4133 CrossRef CAS PubMed.
- Y. Lin, X. Wu, S. Feng, G. Jiang, J. Luo, S. Zhou, L. L. P. Vrijmoed, E. B. G. Jones, K. Krohn, K. Steingrover and F. Zsila, J. Org. Chem., 2001, 66, 6252 CrossRef CAS PubMed.
- X. Wu, X. Liu, G. Jiang, Y. Lin, W. Chan and L. L. P. Vrijmoed, Chem. Nat. Compd., 2005, 41, 27 CrossRef CAS PubMed.
- B. Panda and T. K. Sarkar, J. Org. Chem., 2013, 78, 2413 CrossRef CAS PubMed.
- M. Y. Yamashita, T. Yasumoto, S. Yamada, F. F. A. Bajarias, M. A. Formeloza, M. L. Romero and Y. Fukuyo, Chem. Res. Toxicol., 2004, 17, 1265 CrossRef PubMed.
- L. Brewitz, J. Llaveria, A. Yada and A. Furstner, Chem.–Eur. J., 2013, 19, 4532 CrossRef CAS PubMed.
- (a) A. Furstner, J. Am. Chem. Soc., 2005, 127, 15024 CrossRef PubMed; (b) A. Furstner and C. Aissa, J. Am. Chem. Soc., 2006, 128, 6306 CrossRef PubMed; (c) A. Furstner, E. K. Heilmann and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 4760 CrossRef PubMed.
- S. Das, B. Induvadana and C. V. Ramana, Tetrahedron, 2013, 69, 1881 CrossRef CAS PubMed.
- S. M. Lee, X. F. Li, H. Jiang, J. G. Cheng, S. Seong, H. D. Choi and B. W. Son, Tetrahedron Lett., 2003, 44, 7707 CrossRef CAS PubMed.
- Q. Gao and F. Garcia-Pichel, Nat. Rev. Microbiol., 2011, 9, 791 CrossRef CAS PubMed.
- C. Wang and J. Sperry, Org. Lett., 2011, 13, 6444 CrossRef CAS PubMed.
- (a) R. S. Menon, A. D. Findlay, A. C. Bissember and M. G. Banwell, J. Org. Chem., 2009, 74, 8901 CrossRef CAS PubMed; (b) C. R. Solorio and A. M. Echavarren, J. Am. Chem. Soc., 2010, 132, 11881 CrossRef PubMed; (c) W.-D. Rao, D. Susanti and P. W.-H. Chan, J. Am. Chem. Soc., 2011, 133, 15248 CrossRef CAS PubMed.
- H. Chiba, S. Oishi, N. Fujii and H. Ohno, Angew. Chem., Int. Ed., 2012, 51, 9169 CrossRef CAS PubMed.
- N. Gouault, M. Le Roch, G. de Campos Pinto and M. David, Org. Biomol. Chem., 2012, 10, 5541 CAS.
- (a) G. Dong, T. Xu, B. Yang, X. Lin, X. Zhou, X. Yang and Y. Liu, Chem. Biodiversity, 2011, 8, 740 CrossRef CAS PubMed; (b) N. V. Ivanchina, A. A. Kicha and V. A. Stonik, Steroids, 2011, 76, 425 CrossRef CAS PubMed.
- S. De Marino, M. Iorizzi, F. Zollo, C. D. Amsler, S. P. Greer and J. B. McClintock, Eur. J. Org. Chem., 2000, 4093 CrossRef CAS.
- G. Xiao and B. Yu, Chem.–Eur. J., 2013, 19, 7708 CrossRef CAS PubMed.
- K. C. Nicolaou, G. S. Tria and D. J. Edmonds, Angew. Chem., Int. Ed., 2008, 47, 1780 CrossRef CAS PubMed.
- A. R. Diaz-Marrero, I. Brito, J. M. de La Rosa, J. Darias and M. Cueto, Tetrahedron, 2008, 64, 10821 CrossRef CAS PubMed.
- (a) T. Yoshimitsu, N. Fukumoto, R. Nakatani, N. Kojima and T. Tanaka, J. Org. Chem., 2010, 75, 5425 CrossRef CAS PubMed; (b) T. Umezawa, M. Shibata, K. Kaneko, T. Okino and F. Matsuda, Org. Lett., 2011, 13, 904 CrossRef CAS PubMed; (c) T. Yoshimitsu, R. Nakatani, A. Kobayashi and T. Taneka, Org. Lett., 2011, 13, 908 CrossRef CAS PubMed.
- N. Huwyler and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 13066 CrossRef CAS PubMed.
- O. K. Jeker and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3474 CrossRef CAS PubMed.
- S. Sato, F. Iwata, T. Mukai, S. Yamada, J. Takeo, A. Abe and H. Kawahara, J. Org. Chem., 2009, 74, 5502 CrossRef CAS PubMed.
- B. A. Sobin and F. W. Tanner Jr, J. Am. Chem. Soc., 1954, 76, 4053 CrossRef CAS.
- (a) E. Battaner and D. Vasquez, Biochim. Biophys. Acta, Nucleic Acids Protein Synth., 1971, 254, 316 CrossRef CAS; (b) A. P. Grollman, J. Biol. Chem., 1967, 242, 3226 CAS.
- (a) J. F. Traverse, A. H. Hoveyda and M. L. Snapper, Org. Lett., 2003, 5, 3273 CrossRef CAS PubMed; (b) Y. Hosoya, T. Kameyama, H. Naganawa, Y. Okami and T. Takeuchi, J. Antibiot., 1993, 46, 1300 CrossRef CAS; (c) O. Schwardt, U. Veith, C. Gaspard and V. Joger, Synthesis, 1999, 1473 CrossRef CAS PubMed.
- R. J. Detz, Z. Abiri, R. le Griel, H. Hiemstra and J. H. van Maarseveen, Chem.–Eur. J., 2011, 17, 5921 CrossRef CAS PubMed.
- (a) Common Fragrance and Flavor Materials. Preparation, Properties and Uses, ed. K. Bauer, D. Garbe and H. Surburg, VCH, New York, NY, 1990, 2nd edn Search PubMed; (b) Perspectives in Flavor and Fragrance Research, ed. P. Kraft and K. A. D. Swift, Helvetica Chimica Acta, Zurich, 2005 Search PubMed.
- (a) Y. R. Naves and P. Bachmann, Helv. Chim. Acta, 1945, 28, 1227 CrossRef CAS; (b) P. Kreis and A. Mosandl, Flavour Fragrance J., 1992, 7, 187 CrossRef CAS; (c) M. Kawakami, S. N. Ganguly, J. Banerjee and A. J. Kobayashi, J. Agric. Food Chem., 1995, 43, 200 CrossRef CAS; (d) M. Wust and A. Mosandl, Eur. Food Res. Technol., 1999, 209, 3 CrossRef CAS.
- (a) R. A. Flath and R. R. Forrey, J. Agric. Food Chem., 1977, 25, 103 CrossRef CAS; (b) P. Winterhalter, D. Katzenberger and P. Schreier, Phytochemistry, 1986, 25, 1347 CrossRef CAS.
- N. M. Phuong, T. V. Sung, A. Porzel, J. Schmidt, K. Merzweiler and G. Adam, Phytochemistry, 1999, 52, 1725 CrossRef CAS.
- J. N. Peng, X. Z. Feng, Q. T. Zheng and X. T. Liang, Phytochemistry, 1997, 56, 1119 CrossRef.
- F. Volz, S. H. Wadman, A. Hoffmann-Roder and N. Krause, Tetrahedron, 2009, 65, 1902 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |