Controlled chemistry of tailored graphene nanoribbons for electrochemistry: a rational approach to optimizing molecule detection†
Aída
Martín
a,
Javier
Hernández-Ferrer
b,
Luis
Vázquez
c,
María-Teresa
Martínez
b and
Alberto
Escarpa
*a
aDepartment of Analytical Chemistry, Physical Chemistry and Chemical Engineering, University of Alcalá, E-28871. Alcalá de Henares, Madrid, Spain. E-mail: alberto.escarpa@uah.es; Fax: +34 918854971
bInstituto de Carboquímica ICB-CSIC, C/ Miguel Luesma Castán 4, E-50018 Zaragoza, Spain
cInstituto de Ciencia de Materiales de Madrid (CSIC), C/Sor Juana Inés de la Cruz 3, E-28049 Madrid, Spain
First published on 31st October 2013
Abstract
This work describes a rationalization of the interactions between two fully characterized graphene nanoribbons (GNRs) and a set of significant target molecules. The GNRs were carefully synthesized by unzipping multi-walled carbon nanotubes (MWCNTs) to yield graphene oxide nanoribbons (GNRox) containing 44 wt% oxygen. The GNRox were reduced to yield reduced graphene oxide nanoribbons (GNRred) containing 14 wt%. Each material was characterized by atomic force microscopy, transmission electronic microscopy, Raman spectroscopy, X-ray diffraction, Fourier transform infrared spectroscopy, X-ray photoelectron spectroscopy and voltammetry techniques. Differential pulse voltammetry was used to assess the detection of two strategically selected groups of molecules, including benzenediols, hydroquinone, catechol, and resorcinol, as well as, L-dopa, ascorbic acid, uric acid, and L-tyrosine. The results showed that GNRs provided significantly better electrochemical responses compared to MWCNTs and the non-modified glassy carbon electrode. The chemistry of the few layers of graphene strongly influenced the electrochemical properties of the material. GNRox may be the material of choice for sensing molecules having high oxidation potentials. GNRred, on the other hand, yielded an excellent sensitivity for aromatic molecules in which π–π interactions were dominant or the number of conjugated 1,2-diols present was high. GNRred combines the advantages of the high proportion of sp2-carbon atoms with the presence of a few oxygen moieties remaining in the lattice after the reduction step. The primary interactions responsible for the shift in oxidation potentials were elucidated. This work presents new opportunities for tailoring graphene to a particular sensing application based on the specific chemistry of the molecule.
Introduction
Graphene is a two-dimensional (2-D) sheet of carbon atoms connected by sp2 bonds. The graphene structure conveys extraordinary properties1–3 to the material, such as a high surface area (theoretically 2630 m2 g−1 for single-layer graphene) that is twice the surface area of single-walled carbon nanotubes (SWCNTs). Graphene also shows excellent thermal (k = 5 × 103 W m−1 K−1) and electrical conductivities (σ = 64 mS cm−1). The physical properties of graphene include good optical transparency, a high mechanical strength (Young's modulus, ∼1100 GPa), and a high elasticity. The high surface area, high electrical conductivity, and low production costs, in particular, are of interest for electrochemical applications.Graphenes include graphitic structures that are dimensionally limited to a few to hundred nanometres along the basal plane of the graphene sheets in the x–y plane, such as graphene nanoribbons (GNRs), which can including single- (G-SL), few- (G-FL), or multilayer (G-ML) structures.4 GNRs may be thought of as unzipped carbon nanotubes (CNTs) that have been created with structural control during the unzipping process. The synthesis of GNR5,6 is carried out by applying plasma etching to CNTs embedded in a polymer film,7 by chemically oxidizing CNTs,8,9 by ionic liquid-assisted splitting of CNTs under microwave radiation,10 by the intercalation of metals11 or nanoparticles12 into CNTs, and the posterior exfoliation or unwrapping of MWCNTs using electrical currents and nanomanipulation,13 or by bottom-up strategies14 for producing GNRs of a desired width and length. The main applications15 of GNRs occur in the fields of physics, nanoelectronics, spintronics, and nanoelectromechanical systems (NEMS),16 but GNRs are also relevant to sensing and biosensing applications.
Although the longitudinal unzipping techniques used to synthesize GNRs result in over-oxidation and a plethora of defect sites that do not in general benefit electronic applications,17 these characteristics can actually be useful for certain electrochemical applications, as the authors have proved.
Electrochemical applications using graphene have been extensively discussed in different reviews reported in the literature.18–21
On the other hand, the corresponding applications of GNRs have not been extensively explored. The edge chemistries of chemically functionalized GNRs may offer certain advantages over the edge chemistries of non-functionalized graphene. Non-functionalized graphene presents an inert chemical surface. By contrast, the functional moieties located at the edges of GNRs facilitate the adsorption of molecules by π–π stacking, electrostatic, hydrogen bonding, and covalent interactions.22
A handful of studies have examined the use of GNRs in electrochemical sensing applications. The electrochemical sensing of model electroactive molecules in the presence of reduced GNR-modified screen-printed electrodes has been shown to display a higher sensitivity compared to the sensitivity of a bare screen-printed electrode.17 Various GNR-based sensors and biosensors23–25 have been developed for the detection of urea,26 glucose,27 1-hydroxypyrene,28 cysteine,29 brevetoxin B,30 and 2,4,6-trinitrotoluene.31,32 These sensors displayed excellent electrochemical responses in terms of reproducibility, a low detection limit, and a high selectivity in all cases. In other approaches, GNRs have been incompletely unzipped to develop mixtures of GNR–MWCNTs. The utility of these mixtures for sensing electroactive molecules33 has been examined, yielding good responses.
To explore these features mentioned above and elucidate the chemical interactions between target molecules and graphene in the context of electrochemical sensing, a set of analytically significant target molecules were evaluated using differential pulse voltammetry (DPV). We tested the detection of several target molecules, including three dihydroxybenzene isomers widely used in the chemical industries:34 catechol (CT), resorcinol (RS), and hydroquinone (HQ); the neurotransmitter L-dopa (levodopa/L-3,4 dihydroxyphenylalanine, LD); the amino acid L-tyrosine (L-Tyr); uric acid (UA) and ascorbic acid (AA), which are present in urine and blood serum.35–37 These target molecules are usually oxidized at approximately the same potential; therefore, discrimination among these species in a mixture can be extremely difficult using most solid electrodes.38
In the bibliography interactions between molecules and graphene have been reported using density functional theory and quantum physics.39,40 The studies described here sought to (1) explore the electrochemical performances of fully characterized GNRs with respect to the detection of significant target molecules, and (2) elucidate the chemical interactions between graphenes and the target molecules. Since GNRox were obtained from MWCNTs and the GNRred were obtained from the chemical reduction of GNRox, a reliable and valuable comparison with the critical controls such as the GCE and MWCNTs could be made. Therefore, this traceability is of paramount importance for ascertaining the advantages of GNRs.
Materials and methods
Reagents, standards and samples
The sodium dihydrogen phosphate and disodium hydrogen phosphate used to prepare a phosphate buffered solution (PBS) and the potassium hexacyanoferrate(III) were purchased from Panreac, (Badalona, Spain). LD, UA, RS, HQ, and CT were purchased from Sigma-Aldrich (St. Louis, MO, USA). AA and L-Tyr were obtained from Fluka Chemika (Buchs, UK). The cosmetic sample (Pigmentasa formulation containing 4% (w/w) HQ) was acquired in a local market. Urine samples were recollected from healthy patients.Standard solutions were prepared in 1 mM in 0.1 M phosphate buffered solution (PBS) at pH = 7.4, adjusted using NaOH. All working solutions were protected from light and prepared daily. A 0.1 g (±0.0001) sample of the Pigmentasa formulation was diluted in 10 mL PBS, sonicated in an ultrasonication bath for 10 min, filtered through a 0.2 μm Nylon filter, and diluted 4-fold prior to analysis. Urine samples were diluted 25-fold prior to analysis.
All solutions were prepared with Milli-Q water produced in a Milli-Q system (Millipore, Bedford, MA, USA).
Graphene nanoribbon samples
MWCNTs (0.2% oxygen content) were produced by the arc-discharge method41 in a home-built electric arc-discharge apparatus under standard conditions.42 The MWCNTs were characterized as being straight and highly graphitized.GNRox, 44 wt% oxygen, were synthesized from the MWCNTs via the longitudinal unzipping method in H2SO4/KMnO4.8 These oxidized nanoribbons were used as starting materials to produce GNRred, 14% oxygen, via chemical reduction with N2H4/NH3.43
Apparatus and measurements
Atomic force microscopy (AFM) images were recorded using a Nanoscope IIIa scanning probe microscope (Digital Instruments, USA) operated in the dynamic and contact modes. In both cases, silicon cantilevers (Veeco) with a force constant of ∼40 N m−1 and a nominal radius of 8 nm were employed. The samples used for the measurements were prepared by drop-casting 0.5 μL of the graphene suspension (0.1 mg mL−1) on the surface of a silicon wafer (in the case of the GNRox studies) or on a mica surface (in the case of the GNRred studies). The measurements were obtained in the surroundings of the wet drop where the concentration and aggregation of graphene sheets was lower. Transmission electron microscopy (TEM) images were collected on a JEOL microscope model 2000 FXII at an acceleration potential of 200 kV, which yielded a maximum resolution of 0.28 nm. Raman spectra were obtained on a Micro-Raman confocal spectrophotometer model Horiba Jobin Yvon HR800 UV using a green laser at 532 nm, which yielded a resolution of 0.4 cm−1. X-ray photoelectron spectroscopy (XPS) was carried out on an ESCAPlus Omicron outfitted with a Mg anode and operated at 1253.6 eV with a power of 150 W (14 mA, 10 kV). X-ray diffraction (XRD) measurements were obtained using an X-ray diffractometer Bruker D8 Advance Series. The oxygen content of the graphene samples was direct determined using a Flash 1112 analyzer from Thermo Fisher Scientific.All electrochemical measurements were performed on an electrochemical station μ-AUTOLAB type II (Ecochemie, Utrecht, Holland) using a conventional three-electrode system comprising a platinum wire as an auxiliary electrode, a silver/silver chloride, 3 M KCl (Ag/AgCl) reference electrode (CH Instrument, China), and a glassy carbon electrode (GCE) 3.0 mm in diameter (BAS Instrumental, Warwickshire, UK) as the working electrode. Electrochemical experiments were performed at room temperature.
Procedures
Prior to drop-casting deposition, the GCE was in turn polished using 0.1 and 0.05 μM alumina powders and sequentially sonicated in Milli-Q water and anhydrous ethanol. The GNR and MWCNT-modified electrodes were prepared by casting 10 μL of the GNR solutions (oxide or reduced) or MWCNTs dispersions on the GCE surface (see Table S1†).
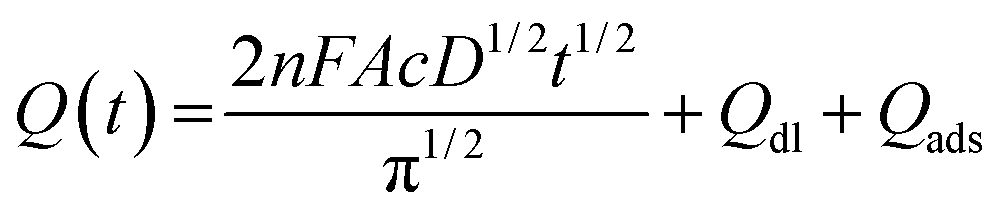 | (1) |
In this equation, A is the effective electrochemical surface area of the working electrode (cm2), c is the concentration of the electroactive species (mol cm−3), n is the number of transfer electrons (that is, 1), F is the Faraday constant, and D is the diffusion coefficient, 7.6 × 10−6 cm2 s−1.46Qdl is the double layer charge, which could be eliminated by background subtraction, and Qads is the Faradaic charge.
DPV was used for the voltammetry analysis with a pulse width of 0.05 V, a pulse frequency of 0.05 s, a pulse cycle of 0.2 s, a pulse interval of 0.004 V, and a standing time of 2 s.
Results and discussion
Characterization of the graphene nanoribbons
The structures and morphologies of the graphene samples were characterized using AFM, as shown in Fig. 1. This figure shows AFM images for a small GNRox sheet with an average thickness of about 0.8 nm and an area of 200 × 150 nm2. The theoretical thickness of a perfectly flat unoxidized sp2-carbon-atom network is predicted to be 0.4 nm.1 The thickness of the GNRox sample measured here was consistent the thickness of two stacked graphene sheets; however, this thickness may also indicate a single layer graphene structure having oxygen functionalities on the surface.47Fig. 1 shows that the GNRred surface was rough due to the presence of stacked small fragments of the reduced graphene sheets. The average thickness of this graphene sample was 1.2 nm, suggesting that the stacked layers extended over an area of 120 × 50 nm2. The GNRred sample displayed a larger number of layers than the graphene oxide sample because the high proportion of sp2-carbons in GNRred increased the extent of π–π stacking interactions.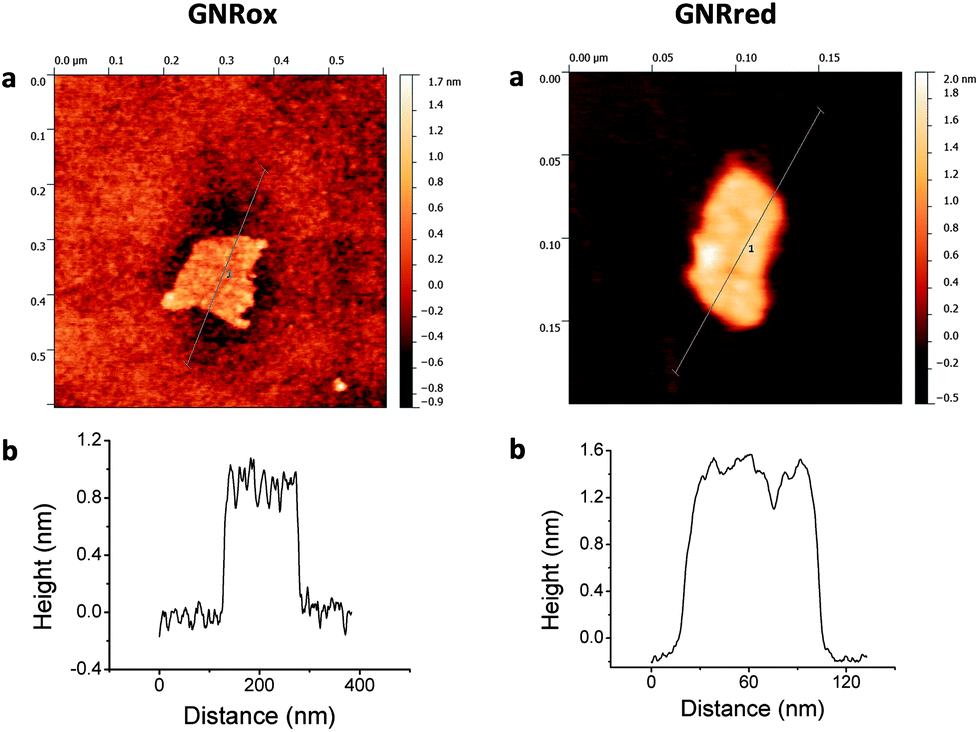 | ||
| Fig. 1 (a) Tapping mode AFM images of GNRox (left) and GNRred (right) on silica and freshly cleaved mica substrates, respectively. (b) Height profiles along the dashed lines indicated in the panels (a). | ||
The graphene morphologies were imaged using TEM, which also demonstrated the successful synthetic process based on the chemical unzipping of MWCNTs. Fig. 2 shows TEM images of a MWCNT sample (A), GNRox (B), and GNRred (C). Although the MWCNTs were more than 1 μm long and had an outer diameter of approximately 12 nm; GNRox displayed stacked layers and folds, and GNRred appeared to form thin layers with folds in the sheets. These micrographs could be used to visualize the openings of the MWCNTs used to generate the graphene layers, as well as the anisotropy of the GNR structures.
 | ||
| Fig. 2 TEM images of the MWCNTs (A), GNRox (B), and GNRred (C) samples. | ||
X-Ray diffraction studies were performed to estimate the average distance between layers in the carbon allotropes. The crystalline structures of graphite, graphene, and carbon nanotubes permit the measurement of the inter-planar spacing and lattice parameters (see ESI, Fig. S2†). The XRD patterns obtained from the MWCNTs presented a peak at 26° corresponding to a basal plane of d002 = 3.34 Å. This peak matched the distance found in the graphite layer structure. The GNRox sample displayed a new peak at 10°, which was attributed to a plane at d001 = 7.33 Å. The separation between layers in the GNRox sample was high due to the presence of oxygen moieties in the lattice. The GNRred peak intensity at 10° was lower and the peak at 26° was higher than the corresponding peak intensities obtained from the GNRox diffractogram. The recovery of sp2-carbon in GNRred facilitated π–π stacking among the layers, and the distance between layers was smaller than the interlayer distance measured in the GNRox sample. XRD studies confirmed the change in the distance between these crystalline structures and the presence of a new graphitic structure.
The Raman spectra of graphite-derived materials usually display a D band at 1360 cm−1 and a G band at 1590 cm−1, and an overtone of the D band occurs at 2650 cm−1 (the 2D or G′ band).7,48 The D band arises from the out-of-plane vibrational modes and is indicative of the number of sp3 carbon atoms present, whereas the G band arises from the presence of in-plane sp2 vibrations. The intensity ratio of the D and G lines (ID/IG ratio), therefore, provides important information about the composition and domains in-plane giving valuable information regarding the average size of the sp2 carbon domains as well.49,50 Fig. S3† illustrates the Raman spectra of the three materials, showing the D, G, and G′ bands. The ID/IG ratio was calculated to be 0.075, 0.52, and 0.66 for the MWCNTs, GNRox, and GNRred samples, respectively. This increase also suggested a decrease in the average size of the sp2 graphitic domains, suggesting that the new graphitic domains created in the GNRred sample were smaller in size but more numerous than in the GNRox sample.48 Because the ID/IG ratio is proportional to the average size of the sp2 carbon domains, a higher ID/IG was attributed to the presence of additional edges (more defects)51 and shorter layers in the GNRred surface, consistent with the AFM results.
XPS was used to study the oxygen content and changes in the sp2–sp3 carbon structure in the graphene layers after chemical reduction of GNRox to obtain GNRred. The presence of sp2-carbon atoms increased significantly and the presence of sp3-carbon and oxygen moieties decreased correspondingly upon reduction of GNRox to GNRred. The presence of carboxyl, carbonyl, alcohol, epoxy, and ether moieties decreased to the same extent. The XPS results revealed the presence of C–N bonds in the as-synthesized GNRred sample as a result of the N2H4/NH3 reduction step (see Fig. S4†). Considering that XPS measurements are sensitive to the elemental composition of the material surface, XPS offers an accurate measure of the oxygen content in the material. Direct determination of oxygen content was directly determined to be 0.2 wt% in the MWCNTs, 44 wt% in the GNRox, and 14 wt% in the GNRred. Clearly, these data differed slightly from the XPS results because the XPS technique is not sensitive to the presence of oxygen groups below the surface and because some of the groups may be lost due to decomposition in the presence of the harsh XPS measurement conditions. The IR spectra were evaluated to corroborate the previous data (see Fig. S5†).
The dispersion of the graphene material was of paramount importance for obtaining these results (see Fig. S1†). Stronger sonication conditions were required to prepare the GNRred dispersion. Because GNRred includes high sp2 content, the material readily stacks to form small piles on an electrode surface that increase the resistivity and reduce the current. The strongest sonication conditions were not necessary to obtain a good GNRox dispersion because the layers did not stack as readily, the material was not found to accumulate, and the signals obtained with or without tip sonication were indistinguishable.
In the last analytical characterization step, the casting electrode was prepared using a 10 μL drop of the GNR suspensions (see Table S1†) and the effective electrochemical surface area was evaluated by chronocoulometry (see Fig. S6†). The estimated areas were 0.030, 0.071, 0.113, and 0.267 cm2 for the non-modified GCE, MWCNTs, GNRox, and GNRred electrodes, respectively. These results revealed that GNRox and GNRred significantly increased the electrochemical surface area of the electrode. This increase corresponded to a 4-fold increase, in the case of GNRox, and a 10-fold increase, in the case of GNRred, over the electrochemical surface area of the bare GCE. The surface areas were higher than the surface area of the MWCNTs by factors of 2 and 5 for the GNRox and GNRred samples, respectively. Good interelectrode precision (n = 3 electrodes) was obtained, with relative standard deviation (RSD) values of 2, 7, and 6% for the MWCNTs, GNRox and GNRred, respectively. These results indicated that the GNRs offered consistently high and reproducible electroactive areas, as predicted.
Interactions between the target molecules and the GNRs based on voltammetry studies
The electrochemical behaviors of the isomers HQ (1,4-diol), CT (1,2-diol), and RS (1,3-diol) were explored on bare GCE, and all the carbon materials (MWCNTs, GNRox, and GNRred) on the GCE, respectively. Fig. 3 top illustrates the electrochemical responses of each electrode to the target molecules. The extraordinarily high oxidation peak currents on the graphene-modified electrodes, as compared to the bare electrode, in the presence of the three target molecules reflected the high conductivity of the GNRs. The high conductivity of the GNRred electrode was particularly remarkable as a result of the high sp2-carbon content, which increased the electrical conductivity of this material. As an example, the electrochemical response of GNRred to catechol was one order of magnitude higher than the response to HQ, (see Table S2†). The presence of incompletely unzipped MWCNTs, if any, was not expected to significantly affect the detection performance because the MWCNT electrode displayed a low signal level.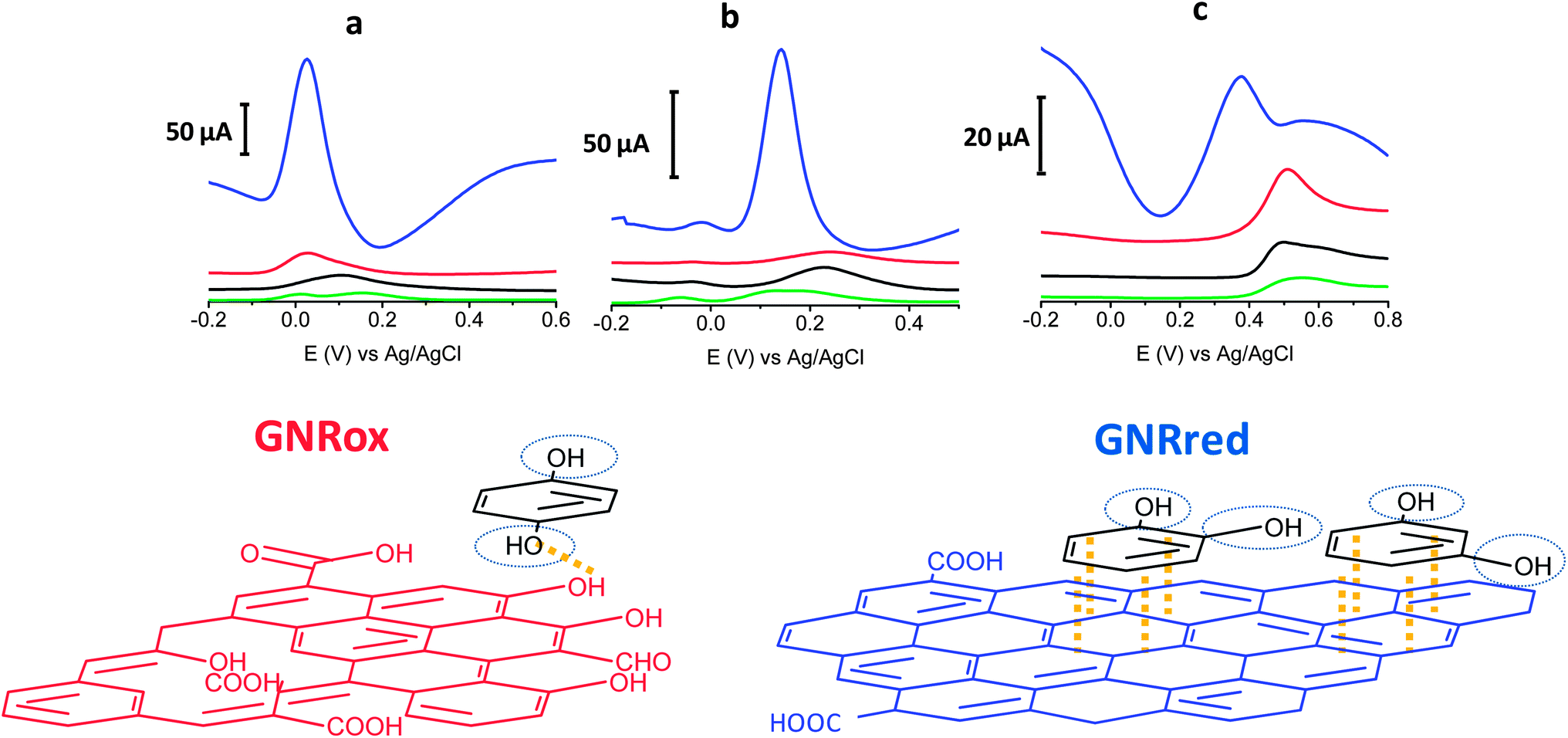 | ||
Fig. 3 (Top) Differential pulse voltammograms obtained from 1 mM HQ (a), CT (b), and RS (c). ( ) Bare GCE, ( ) Bare GCE, ( ) MWCNTs, ( ) MWCNTs, ( ) GNRox, and ( ) GNRox, and ( ) GNRred. (See the Experimental section for a description of the working conditions). (Bottom) Schematic diagram of the interactions between the target molecules (black) and the graphene materials which showed the lower oxidation potential. The oxidation reaction center is indicated by the blue circles and the main interactions are indicated in yellow. ) GNRred. (See the Experimental section for a description of the working conditions). (Bottom) Schematic diagram of the interactions between the target molecules (black) and the graphene materials which showed the lower oxidation potential. The oxidation reaction center is indicated by the blue circles and the main interactions are indicated in yellow. | ||
The chemistry underlying the shifts in oxidation potentials in the electrochemical detection of the target molecules may be understood as follows. 1,4-Benzenediol readily oxidizes at low potentials to produce 1,4-benzoquinones, whereas the oxidation of 1,3-diol is less favorable due to the lack of conjugation, thereby preventing the formation of the benzoquinone, as it occurs with 1,4 and 1,2-benzenediols. Fig. 3 bottom illustrates the possible interaction processes between the target molecules and the graphene material. We hypothesized that the availability of more π–π interactions in GNRred than in GNRox facilitated the electrocatalysis of CA and RS and, as a consequence, facilitated oxidation of both benzenediols with GNRred. By contrast, 1,4-benzenediol displayed similar oxidation potentials on both GNRs, suggesting that hydrogen bonds dominated the interactions between the oxide moieties on the edges of the GNR surfaces and the molecules, due to the similar potential of the HQ in the presence of GNRred and GNRox.
Fig. 4 top presents the detection results of LD, AA, L-Tyr, and UA on GCE, MWCNTs, GNRox, and GNRred. A comparison of the graphene modified electrodes and the GCE and MWCNTs controls revealed that both graphene electrodes displayed significantly higher current peaks compared to the MWCNT control. The results obtained from GNRred were particularly spectacular. The exceptional electrocatalytic properties exhibited by this material enabled the detection of AA, even below 0 V. Moreover, GNRred displayed analytical response from 5 to 10-fold higher compared to the response of the GCE.
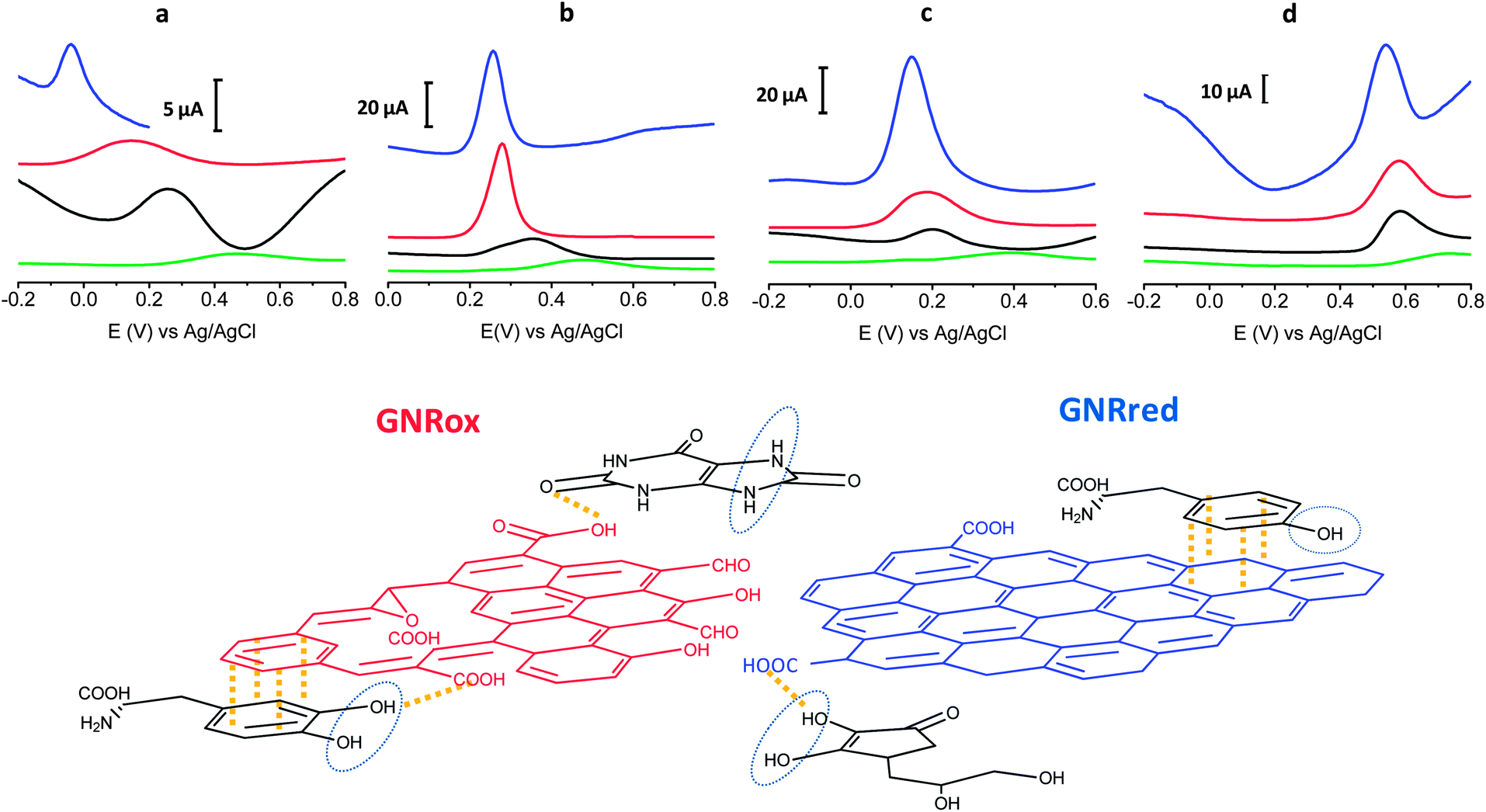 | ||
| Fig. 4 (Top) Differential pulse voltammograms for 1 mM AA (a), LD (b), UA (c) and L-Tyr (d). () Bare GCE, () MWCNTs, () GNRox and () GNRred. (See Experimental section for working conditions). (Bottom) Schematic diagram illustrating the interactions between the analytes (black) and graphene surfaces which showed the lower oxidation potential. The oxidation reaction centers are indicated by the blue circle and the main interactions are indicated in yellow. | ||
Table S3† summarizes the oxidation potentials and analytical signals obtained during the DPV measurements for the four molecules.
The enhanced sensitivity of the GNRred electrode was attributed to the good conductivity of the material. These results may be understood in terms of the previous results obtained in analytical characterization studies, which identified a high electrochemical surface area and the presence of high sp2-carbon content. Fig. 4 bottom provides a schematic diagram illustrating our rationalization of the interactions between the GNR and the target molecules. The oxygen groups of the GNRox layers and those oxygen functionalities remaining in the GNRred lattice (this latter material supposed to be in the form of carboxylic acids and carbonyls, as described in the model proposed by Lerf–Klinowski43,52,53) are expected to interact with the target molecules via hydrogen bonds. In the detection of AA, GNRred yielded the strongest electrocatalysis compared to UA, where similar electrocatalytic effect was found for both graphenes. This shift could be explained considering interactions with the oxidation center of AA and GNRred and without this oxidation center in the case of UA.
LD, which basic structure derived from CT, displayed indistinguishable behavior for GNRred and GNRox. Therefore, the predominant π–π interactions plus hydrogen interactions between the benzenediol group and the GNRred and GNRox surfaces appeared to be responsible for the similar responses.
The oxidation process of L-Tyr was more difficult due to the presence of only one hydroxyl group. For this reason the similar oxidation potentials observed on all carbon materials (MWCNTs, GNRox, and GNRred) could be explained in terms of weak π–π interactions between the tyrosine and the carbon materials.
Fig. 5A shows the electrochemical responses of mixtures of the three isomers: HQ, CT, and RS on GCE, MWCNTs, GNRox, and GNRred. The responses revealed that although the bare GCE did not permit the simultaneous detection of the benzene 1,2 and 1,4-diols, the electrocatalytic properties of GNRox and GNRred allowed the simultaneous and separate detection of the three target molecules. Fig. 5B shows that the selective detection of LD, AA, and UA could only be achieved using the GNRred electrode. Fig. 5C shows that the separate components of a mixture comprising tyrosine and UA could be identified using the GNRred electrode as well. The detection of individual target molecules in a mixture relies on the availability of distinct interactions between each nanomaterial and target molecule, and consequently, to the exceptional electrocatalytic properties exhibited.
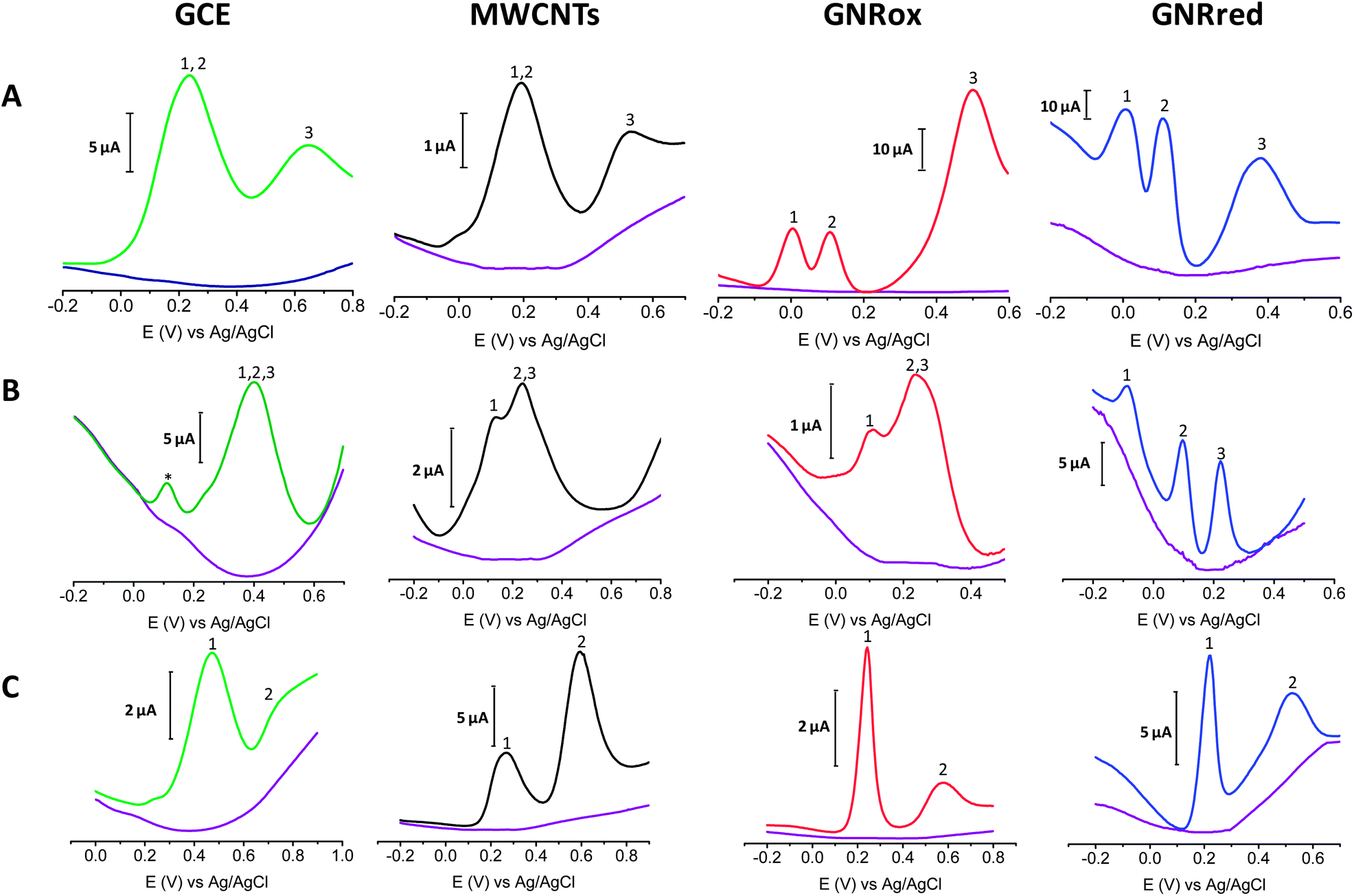 | ||
Fig. 5 (A) Voltammograms for the detection of: HQ 0.25 mM (peak 1); CA 0.25 mM (peak 2) and RS 1.0 mM (peak 3). (B) AA 0.5 mM (peak 1); LD 0.05 mM (peak 2), and UA 0.05 mM (peak 3). (C) UA 1.0 mM (peak 1) and L-Tyr 1.0 mM (peak 2). ( ) Background signal, () bare GCE, () MWCNTs, () GNRox, and () GNRred. Note: * pre-peak oxidation in LD. (See the Experimental section for a description of the working conditions.) ) Background signal, () bare GCE, () MWCNTs, () GNRox, and () GNRred. Note: * pre-peak oxidation in LD. (See the Experimental section for a description of the working conditions.) | ||
The strength of the voltammetric studies was examined by measuring the technique's repeatability and reproducibility. The repeatability (multiple experiments conducted on the same day) and the reproducibility (multiple experiments conducted on different days) of the oxidation peak positions was found to be excellent, with RSD values of <4% (n = 10). Good inter-electrode precision was achieved, with RSD values of <6% (n = 5, same day) and RSD < 9% (n = 5, different days), for the GNRred electrode. The precision was significantly better than the precision obtained from the GCE electrode (RSD < 10% n = 4 electrodes, same day).
It is worth noting that real samples were also tested. Fig. S7† illustrates the electrochemical detection of HQ and UA in cosmetic and urine samples, respectively. Interestingly, in both cases, the GNRs displayed better intensity currents than the GCE. The excellent results in complex matrix suggest that this technique is suitable for the analysis of complex samples. Good precision was achieved for both samples. The cosmetic sample analysis was characterized by an RSD of <0.5% for the oxidation peaks and an RSD of <2% for the peak currents. The urine sample analysis was characterized by an RSD of <0.5% for the oxidation peaks and an RSD of <10% for the peak currents.
Conclusions
Both graphenes displayed excellent electrochemical behaviour in the detection of the target molecules, being this behaviour rigorously attribute to graphene and not to other graphitic materials. The interactions between the target molecules and the GNR materials present new opportunities in the field of electrochemistry. A suitable graphene material may potentially be tailored for a particular detection application with consideration for the relevant degree of oxidation and sp2 structure in the electrode that would be required to promote hydrogen bonding or π–π interactions between the electrode and the target molecule. GNRox appeared to be ideal for the detection of molecules having high oxidation potentials, whereas GNRred displayed a better response to aromatic molecules, such as the conjugated 1,2-diols, which interacted with the electrode predominantly through π–π interactions.Although both graphenes exhibited excellent electrochemical performance, GNRred became an exceptional material. The completely opened GNRred lattices, which displayed an average thickness of 1.2 nm and a high percentage of sp2-carbon were obtained through the synthetic methods described here. GNRred exhibited a 10-fold higher electrochemical surface area and a better analytical performance in the context of electrochemical sensing in comparison with the GCE. These GNRred features allowed for improved electrochemical sensing and suggested that this material was suitable for the electrochemical detection of target molecules. The outstanding electrocatalytic effect performance relies on the presence of a restored sp2 structure that includes oxygen groups in the GNRred lattice. These combined features yielded excellent electrocatalytic properties due to the effects of both the π–π and hydrogen bonding interactions between the molecules and the GNRs. These interactions enhanced electrocatalysis at the primary catalytic sites at the oxidation centers. The studies described here demonstrate that GNRred is a promising material for use in molecular sensing, with very rich chemistry and electrochemistry properties. GNRred combines the advantages derived from both the high proportion of sp2-carbon atoms available within the surface layers (similar to the structure of exfoliated graphene, 0.4 nm corresponds to one layer) with the advantages derived from the remaining oxygen moieties present on the surface. The results presented here open new opportunities for electrochemical sensing applications and guide the process of tailoring a suitable graphene electrode material for use in a particular molecular detection application.
Acknowledgements
Financial support provided by the Spanish Ministry of Science and Innovation (CTQ2011-28135), the Spanish ministry of Economy and Competitiveness (FIS2012-38866-C05-05, MICINNTEC2010-15736), as well as by the AVANSENS program from the Community of Madrid (P2009/PPQ-1642) is gratefully acknowledged. D. Aída Martín acknowledges the FPU fellowship received from the Ministry of Education, Culture and Sports. J. Hernández-Ferrer acknowledges the Spanish Superior Council for Scientific Research (CSIC) for his JAE-Doc contract.Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- M. Terrones, Nature, 2009, 458, 845–846 CrossRef CAS PubMed.
- L. Ma, J. Wang and F. Ding, ChemPhysChem, 2013, 14, 47–54 CrossRef CAS PubMed.
- L. Jiao, L. Zhang, X. Wang, G. Diankov and H. Dai, Nature, 2009, 458, 877–880 CrossRef CAS PubMed.
- D. V. Kosynkin, A. L. Higginbotham, A. Sinitskii, J. R. Lomeda, A. Dimiev, B. K. Price and J. M. Tour, Nature, 2009, 458, 872–876 CrossRef CAS PubMed.
- C. K. Chua, Z. Sofer and M. Pumera, Chem.–Asian J., 2012, 7, 2367–2372 CrossRef CAS PubMed.
- S. Vadahanambi, J. Jung, R. Kumar, H. Kim and I. Oh, Carbon, 2013, 53, 391–398 CrossRef CAS PubMed.
- A. G. Cano-Marquez, F. J. Rodriguez-Macias, J. Campos-Delgado, C. G. Espinosa-Gonzalez, F. Tristan-Lopez, D. Ramirez-Gonzalez, D. A. Cullen, D. J. Smith, M. Terrones and Y. I. Vega-Cantu, Nano Lett., 2009, 9, 1527–1533 CrossRef CAS PubMed.
- L. Ci, Z. Xu, L. Wang, W. Gao, F. Ding, K. F. Kelly, B. I. Yakobson and P. M. Ajayan, Nano Res., 2008, 1, 116–122 CrossRef CAS PubMed.
- K. Kim, A. Sussman and A. Zettl, ACS Nano, 2010, 4, 1362–1366 CrossRef CAS PubMed.
- X. Yang, X. Dou, A. Rouhanipour, L. Zhi, H. J. Raeder and K. Muellen, J. Am. Chem. Soc., 2008, 130, 4216–4217 CrossRef CAS PubMed.
- M. Terrones, A. R. Botello-Mendez, J. Campos-Delgado, F. Lopez-Urias, Y. I. Vega-Cantu, F. J. Rodriguez-Macias, A. L. Elias, E. Munoz-Sandoval, A. G. Cano-Marquez, J. Charlier and H. Terrones, Nano Today, 2010, 5, 351–372 CrossRef PubMed.
- Y. Guo, W. Guo and C. Chen, Appl. Phys. Lett., 2008, 92, 243101 CrossRef.
- D. B. Shinde, J. Debgupta, A. Kushwaha, M. Aslam and V. K. Pillai, J. Am. Chem. Soc., 2011, 133, 4168–4171 CrossRef CAS PubMed.
- M. Pumera, A. Ambrosi, A. Bonanni, E. L. K. Chng and H. L. Poh, TrAC-Trends Anal. Chem., 2010, 29, 954–965 CrossRef CAS PubMed.
- D. A. C. Brownson and C. E. Banks, Analyst, 2010, 135, 2768–2778 RSC.
- Y. Shao, J. Wang, H. Wu, J. Liu, I. A. Aksay and Y. Lin, Electroanalysis, 2010, 22, 1027–1036 CrossRef CAS.
- S. Wu, Q. He, C. Tan, Y. Wang and H. Zhang, Small, 2013, 9, 1160–1172 CrossRef CAS PubMed.
- S. Zhang, S. Tang, J. Lei, H. Dong and H. Ju, J. Electroanal. Chem., 2011, 656, 285–288 CrossRef CAS PubMed.
- F. Valentini, D. Romanazzo, M. Carbone and G. Palleschi, Electroanalysis, 2012, 24, 872–881 CrossRef CAS.
- X. Dong, Q. Long, J. Wang, M. B. Chan-Park, Y. Huang, W. Huang and P. Chen, Nanoscale, 2011, 3, 5156–5160 RSC.
- F. Valentini, M. Carbone and G. Palleschi, Anal. Bioanal. Chem., 2013, 405, 3449–3474 CrossRef CAS PubMed.
- Y. Yang, J. Zhou, H. Zhang, P. Gai, X. Zhang and J. Chen, Talanta, 2013, 106, 206–211 CrossRef CAS PubMed.
- R. K. Srivastava, S. Srivastava, T. N. Narayanan, B. D. Mahlotra, R. Vajtai, P. M. Ajayan and A. Srivastava, ACS Nano, 2012, 6, 168–175 CrossRef CAS PubMed.
- X. Shen, Y. Cui, Y. Pang and H. Qian, Electrochim. Acta, 2012, 59, 91–99 CrossRef CAS PubMed.
- S. Wu, X. Lan, F. Huang, Z. Luo, H. Ju, C. Meng and C. Duan, Biosens. Bioelectron., 2012, 32, 293–296 CrossRef CAS PubMed.
- J. Tang, L. Hou, D. Tang, J. Zhou, Z. Wang, J. Li and G. Chen, Biosens. Bioelectron., 2012, 38, 86–93 CrossRef CAS PubMed.
- M. S. Goh and M. Pumera, Anal. Bioanal. Chem., 2011, 399, 127–131 CrossRef CAS PubMed.
- S. M. Tan, C. K. Chua and M. Pumera, Analyst, 2013, 138, 1700–1704 RSC.
- C. Sun, C. Chang, H. Lee, J. Zhou, J. Wang, T. Sham and W. Pong, ACS Nano, 2011, 5, 7788–7795 CrossRef CAS PubMed.
- S. Suresh, V. Srivastava and I. M. Mishra, Int. J. Electr. Electron. Eng., 2012, 3, 32 Search PubMed.
- J. M. Zen, C. T. Hsu, Y. L. Hsu, J. W. Sue and E. D. Conte, Anal. Chem., 2004, 76, 4251–4255 CrossRef CAS PubMed.
- A. Salimi, H. MamKhezri and R. Hallaj, Talanta, 2006, 70, 823–832 CrossRef CAS PubMed.
- K. Reddaiah, T. M. Reddy and P. Raghu, J. Electroanal. Chem., 2012, 682, 164–171 CrossRef CAS PubMed.
- R. D. Oneill, Analyst, 1994, 119, 767–779 RSC.
- Z. Wang, H. Hu, Y. Wei and Q. Huang, Phys. B, 2010, 405, 3895–3898 CrossRef CAS PubMed.
- M. Roos, D. Kuenzel, B. Uhl, H. Huang, O. B. Alves, H. E. Hoster, A. Gross and R. J. Behm, J. Am. Chem. Soc., 2011, 133, 9208–9211 CrossRef CAS PubMed.
- M. V. Antisari, R. Marazzi and R. Krsmanovic, Carbon, 2003, 41, 2393–2401 CrossRef.
- A. M. Benito, W. K. Maser and M. T. Martinez, Int. J. Nanotechnol., 2005, 2, 71–89 CAS.
- X. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2010, 114, 832–842 CAS.
- F. C. Anson, Anal. Chem., 1964, 36, 932–934 CrossRef CAS.
- F. C. Anson and R. A. Osteryoung, J. Chem. Educ., 1983, 60, 293–296 CrossRef CAS.
- N. P. C. Stevens, M. B. Rooney, A. M. Bond and S. W. Feldberg, J. Phys. Chem. A, 2001, 105, 9085–9093 CrossRef CAS.
- M. Zhou, Y. Zhai and S. Dong, Anal. Chem., 2009, 81, 5603–5613 CrossRef CAS PubMed.
- A. Ambrosi, A. Bonanni, Z. Sofer, J. S. Cross and M. Pumera, Chem.–Eur. J., 2011, 17, 10763–10770 CrossRef CAS PubMed.
- E. B. Barros, K. Sato, G. G. Samsonidze, A. G. Souza Filho, M. S. Dresselhaus and R. Saito, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 245435 CrossRef.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- M. Cheng, R. Yang, L. Zhang, Z. Shi, W. Yang, D. Wang, G. Xie, D. Shi and G. Zhang, Carbon, 2012, 50, 2581–2587 CrossRef CAS PubMed.
- A. Lerf, H. Y. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- H. Y. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53–56 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra44235g |
| This journal is © The Royal Society of Chemistry 2014 |