
A microfluidic device for multiplex single-nucleotide polymorphism genotyping†
Abstract
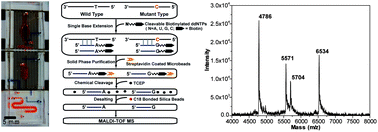
Single-nucleotide polymorphisms (SNPs) are the most abundant type of genetic variations; they provide the genetic fingerprint of individuals and are essential for genetic biomarker discoveries. Accurate detection of SNPs is of great significance for disease prevention, diagnosis and prognosis, and for prediction of drug response and clinical outcomes in patients. Nevertheless, conventional SNP genotyping methods are still limited by insufficient accuracy or labor-, time-, and resource-intensive procedures. Microfluidics has been increasingly utilized to improve efficiency; however, the currently available microfluidic genotyping systems still have shortcomings in accuracy, sensitivity, throughput and multiplexing capability. To address these challenges, we developed a multi-step SNP genotyping microfluidic device, which performs single-base extension of SNP specific primers and solid-phase purification of the extension products on a temperature-controlled chip. The products are ready for immediate detection by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS), providing identification of the alleles at the target loci. The integrated device enables efficient and automated operation, while maintaining the high accuracy and sensitivity provided by MS. The multiplex genotyping capability was validated by performing rapid, accurate and simultaneous detection of 4 loci on a synthetic template. The microfluidic device has the potential to perform automatic, accurate, quantitative and high-throughput assays covering a broad spectrum of applications in biological and clinical research, drug development and forensics.
 Please wait while we load your content...
Please wait while we load your content...

