
Dendrimer-based multilayer nanocarrier for potential synergistic paclitaxel–doxorubicin combination drug delivery†
Abstract
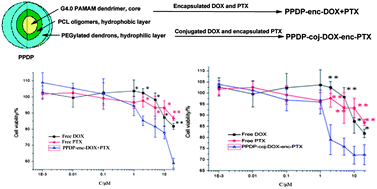
A unimolecular micelle-like nanocarrier (PPDP) based on poly(amidoamine) (PAMAM) dendrimers was synthesized to develop combined drug delivery systems for intensifying the therapeutic effect on tumors. The carrier was designed as a dual drug container with oligo-poly(ε-caprolactone) (PCL) and PEGylated amidoamine dendrons as the hydrophobic and hydrophilic domains for each molecule. PPDP was characterized with a size of 10–20 nm, indicating that it mostly remained in the single molecular state rather than in aggregates. Based on PPDP, two drug delivery systems of PPDP-enc-DOX + PTX and PPDP-coj-DOX-enc-PTX were constructed. PPDP-enc-DOX + PTX was prepared by physically encapsulating doxorubicin (DOX) and paclitaxel (PTX) in the different layers of the PPDP, while PPDP-coj-DOX-enc-PTX was fabricated by conjugating DOX through the pH-sensitive linkage on the exterior and embedding PTX in the interior of the PPDP. The drug loading or conjugating and the drug release behaviors were tested. The cytotoxicity, the cell uptake and the intracellular distribution of PPDP-enc-DOX + PTX and PPDP-coj-DOX-enc-PTX were measured in vitro. For both of the systems, the release of PTX was achieved in a sustained manner during 48 h in vitro, while for the PPDP-coj-DOX-enc-PTX system, the liberation of DOX could be controlled. PPDP-enc-DOX + PTX and PPDP-coj-DOX-enc-PTX both showed the ability to simultaneously deliver two drugs into the same tumor cells and showed higher cytotoxicity toward MCF-7/ADR and MCF-7 cells than free DOX or PTX alone due to the synergistic effect of the drugs. Notably, PPDP-enc-DOX + PTX showed a better inhibition effect on MCF-7/ADR cells as it could deliver DOX into the nucleus, while PPDP-coj-DOX-enc-PTX had the stronger cytotoxicity to MCF-7 cells due to the steady sustained release manner of the two drugs.
 Please wait while we load your content...
Please wait while we load your content...

