
Multicomponent hybrids with surfactant-encapsulated europium polyoxometalate covalently bonded ZnO and tunable luminescence
Abstract
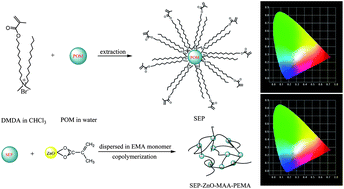
In this paper, europium polyoxometalate (Na9EuW10O36·32H2O, abbreviated as POM) is encapsulated into the surfactant dodecyl(11-(methacryloyloxy)undecyl) dimethylammonium bromide (DMDA) to form surfactant-encapsulated polyoxometalate (SEP). Methacrylic group-modified ZnO nanoparticles (designated ZnO-MAA) are also prepared through a sol–gel reaction between zinc methacrylate and LiOH. Finally, the synthesized SEP, ZnO-MAA and three different ester units (ethyl methacrylate (EMA), 2-hydroxyethyl methacrylate (HEMA) and 2,2,3,4,4,4-hexafluorobutyl methacrylate (HFMA)) are covalently bonded through a copolymerization reaction in the presence of benzoyl peroxide (BPO). A detailed physical characterization and especially the photoluminescence of these hybrid materials are studied in detail. The results provide a method to assemble multicomponent hybrid materials with europium polyoxometalate, a semiconductor compound and polymer unit, whose luminescence can be tuned and integrated.
 Please wait while we load your content...
Please wait while we load your content...

