
Flavonoids from Achyrocline satureioides: promising biomolecules for anticancer therapy
Abstract
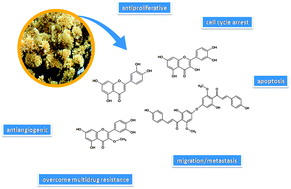
Cancer is the major public health problem worldwide; consequently, the search for new chemotherapeutic drugs is constant. Most of these agents are derived from natural sources, which are the major consistent basis for the search for modern anticancer medicines. In this context, numerous studies indicate flavonoids as a potential new class of secondary metabolites for anticancer therapy. In this review, special attention was addressed to flavonoids present in Achyrocline satureioides, a widely used medicinal plant with several and a well established range of biological properties. Two of these flavonoids are extensively studied for anticancer therapy, quercetin and luteolin, followed by 3-O-methylquercetin. Achyrobichalcone, recently isolated from A. satureioides by our group, can also represent a promising chemotherapeutic biomolecule due to its similarity with other cytotoxic bichalcones to cancer cell lines. The anticancer properties of these flavonoids, specially quercetin and luteolin, type of cell death, mechanisms and molecular targets involved were described. In general, these effects were observed due to the inhibition of cell proliferation, cell cycle arrest, apoptosis, inhibition of angiogenesis, prevention of migration/metastasis and overcoming multidrug resistance, alone or in combination with commonly used chemotherapeutic drugs. All these successful findings in preclinical studies suggest that these flavonoids are promising biomolecules for the development of new anticancer drugs in the future.
 Please wait while we load your content...
Please wait while we load your content...

