Selective continuous flow extractive denitrogenation of oil containing S- and N-heteroaromatics using metal-containing ionic liquids supported on monolithic silica with hierarchical porosity†
Paulo
Forte
a,
Alexander
Sachse
b,
Michael
Maes
a,
Anne
Galarneau
b and
Dirk
De Vos
*a
aCentre for Surface Chemistry and Catalysis, Katholieke Universiteit Leuven, Kasteelpark Arenberg 23, B-3001 Leuven, Belgium. E-mail: dirk.devos@biw.kuleuven.be; Fax: +32 16 321998; Tel: +32 16 321639
bInstitut Charles Gerhardt Montpellier, UMR 5253, CNRS-UM2-ENSCM-UM1, ENSCM, 8 rue de l'École Normale, 34296 Montpellier Cedex 5, France
First published on 28th October 2013
Abstract
The removal of heteroaromatic nitrogen and sulfur impurities from a model oil through extraction with ionic liquids (ILs) containing metal salts was performed in view of the purification of fuel feeds. Chloride and bis(trifluoromethanesulfonyl)imide salts of Cu+, Cu2+ and Fe3+ were used. The systems based on ILs and metal compounds were applied both in batch-like liquid–liquid and continuous flow liquid–solid conditions. In a first phase, liquid–liquid biphasic extraction was used to choose the most adequate IL classes; the suitable systems were immobilized on a solid support to form metal-containing supported ionic liquid phases (SILPs). In a next phase, these SILPs were applied in breakthrough experiments. A selective extraction of N-compounds was achieved with metal-containing ionic liquids, in both liquid–liquid and liquid–solid conditions. The breakthrough experiments using Cu(NTf2)2− and FeCl4−-containing [BMIM][NTf2] SILPs immobilized on hierarchically structured silica monoliths resulted in an efficient separation of all the nitrogen compounds from the other impurities in the model oil.
Introduction
The ever rising consumption rate of fossil fuels implies that sweet crude sources are depleted at a fast rate and heavier and dirtier crude sources need to be exploited. This means that the fuel feedstocks will increasingly contain heavier components and higher concentrations of pollutant impurities, such as heteroaromatic nitrogen and sulfur compounds. On the other hand, recent legislation has steadily set the bar lower regarding the legal maximum concentration of sulfur impurities in fuel feeds. European specifications, for example, demand a total S concentration down to as low as 10 ppm.1The current industrial staples for the deep desulfurization of fuel feeds are through reductive and/or oxidative processes. The reductive processes, called hydrodesulfurization (HDS), often employ polymetallic clusters of Co, Mo and/or Ni.1a,2 It is known that HDS is less efficient in the removal of the heavier and more sterically hindered sulfur heteroaromatics such as benzothiophene (BT) or dibenzothiophene (DBT). These compounds require higher pressures of hydrogen and overall harsher reaction conditions.1a,2b,3 The oxidative desulfurization (ODS) methods currently employ such catalysts as organic acids,4 polyoxometalates,4a,5 titanium-containing zeolites and silicates,6 FeIII salts,7etc and produce ultra-low sulfur diesel by using cheap oxidants such as H2O2 or O2 in mild conditions.8 However, both reductive and oxidative processes are inhibited by the presence of nitrogen impurities,1a,9 due to adsorption of these compounds – or their reduction or oxidation products – on the catalytic sites. Since nitrogen compounds are less reactive than their sulfur counterparts, their removal through catalytic hydrodenitrogenation is considerably more difficult. All these factors make that the selective denitrogenation of fuel feeds in the presence of sulfur impurities poses a major technological challenge.
The use of ionic liquids (ILs) for desulfurization of fuel feeds has been widely researched.1b,10 Their broad liquid ranges and negligible volatility present obvious engineering advantages while, from the chemical point of view, their immiscibility with fuel oils and affinity for the target impurities can be systematically fine-tuned. Commonly, their use has been as extracting phase for the biphasic extraction of heteroaromatic impurities.10,11 ILs have also been found to be excellent solvents for homogeneous catalysts in biphasic conditions.12 The presence of metal catalysts dissolved or embedded in the structure of the IL itself has made IL systems also suitable for use in ODS processes.13 The use of ILs in which ionic metal species are an intrinsic part of the structure has mostly been investigated for simple extractive purification, as in the case of the anionic halide complexes of aluminum(III),11b,h,14 zinc(II),11d,14,15 copper(I),16 copper(II)14 and iron(III).11e,14 The use of this type of ILs has, however, limitations: tetrachloroaluminate species are air- and moisture-sensitive, and anionic metal halides in general are very corrosive, which limits their use in bulk quantities. The promising characteristics of ionic liquids combined with the limitations of the current techniques highlighted above result in a rising demand for the development of more benign metal-containing IL systems for the selective denitrogenation of fuel feeds.
Until now, most results on extractive purification of fuels using ionic liquids were obtained with liquid–liquid biphasic, batch-like processes. This implies regeneration of the IL phase by either distillation or back-extraction before proceeding to the next batch. The immobilization of an IL on a solid support to form a supported ionic liquid phase (SILP) affords many advantages relative to classical liquid–liquid batch conditions.17 The charged groups allow a very efficient immobilization by adsorption to polar supports while maintaining the functional polar or apolar groups that confer the appropriate properties for the desired selectivity. The well-studied chemistry of biphasic IL-solvent systems is then improved by the increase in the surface area available to the substrate, which not only increases the activity of the IL as catalytic or extracting phase but also minimizes one of the greatest drawbacks in the use of ILs, viz. the slow diffusion caused by their high viscosity.18 The adsorptive immobilization of ILs also presents, over covalent binding, the advantage of not requiring the chemical modification of the ILs and/or the metallic species' structure. The desulfurization of liquid fuel feeds using SILPs has been reported, whereby the SILP containing a metal halide/IL mixture is stirred in contact with the fuel, in a batch-like process.15 Besides the previously discussed disadvantages of metal halide/IL systems, these existing studies also do not address the problem of the selective denitrogenation necessary for the success of other, more efficient desulfurization processes such as HDS or ODS. Moreover, the choice for a non-continuous process implies that regeneration steps are necessary.
The application of flow-through methodology has been increasingly adopted in multiphasic processes which require the use of solid supports.19 Packed-bed columns with particulate supports have been used in industrial processes but technically, the handling of particulates is regarded more and more as unwanted by the industry, due to health and safety concerns. Furthermore, the possible formation of preferential elution pathways and the broad distribution of residence times may lead to poor process efficiencies and reproducibility. The use of hierarchically-structured macroporous/mesoporous monolithic supports presents a very attractive alternative.20 The homogeneity of their pore structure, the high available surface area and the ease of their synthesis, fitted to the reactor shape, eliminate the drawbacks of packed-bed reactors. Silica-based monoliths have inherent advantages over polymer- or zeolite-based ones: their ease of synthesis affords a wide range in macropore size, and they are chemically, mechanically and thermally stable. These silica monoliths present a hierarchical structure consisting of a highly interconnected, homogeneous network of macropores of about 2 to 20 μm of diameter with mesopores from 5 to 15 nm. Our group has already reported the applications of monolithic supports in fine chemical synthesis21 and metal extraction from effluents.22 Because of their polar nature and the presence of the macropore network, they are especially suited to the use with adsorption-immobilized SILPs.23
The present work aims at the study of the selective extractive denitrogenation of fuel feeds containing heavy heteroaromatic N and S impurities by supported ionic liquid phase systems containing bistriflimide or halide metal salts immobilized on hierarchically structured silica monoliths.
Results and discussion
To mimic the typical heavy N and S impurities usually found in fuel feeds, a model oil was prepared. This was a heptane/toluene (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio) mixture with a total impurity concentration of 7.4 × 10−3 M, with 4.6 × 10−3 M of nitrogen compounds and 2.8 × 10−3 M of sulfur compounds. The N-compounds tested were indole (I), 1,2-dimethylindole (DMI) and N-methylcarbazole (NMC) while the S-compounds were benzothiophene (BT) and dibenzothiophene (DBT) (Scheme 1).
:1 volume ratio) mixture with a total impurity concentration of 7.4 × 10−3 M, with 4.6 × 10−3 M of nitrogen compounds and 2.8 × 10−3 M of sulfur compounds. The N-compounds tested were indole (I), 1,2-dimethylindole (DMI) and N-methylcarbazole (NMC) while the S-compounds were benzothiophene (BT) and dibenzothiophene (DBT) (Scheme 1).
 | ||
| Scheme 1 Structures of the N and S impurities studied in this work. | ||
The IL systems suitable for use in continuous setups were developed in two phases. In a first phase, a set of preliminary batch-type experiments were run to choose the most appropriate IL system for the selective extraction of nitrogen compounds from the model oil.
Biphasic liquid–liquid extractions
The initial tests were performed as biphasic liquid–liquid extraction experiments, subjecting the N- and S-containing model oil to contact with an ionic liquid phase in a sealed vial while stirring. Scheme 2 shows the structures of the cations and anions of the ionic liquids used at this stage. | ||
| Scheme 2 Molecular structures of the ILs used in the biphasic experiments. | ||
Ionic liquids and ionic liquid mixtures
The heterocyclic structures have an important effect on the uptakes, and indole, as a N-heterocycle with an available N–H bond, is clearly the preferred compound in practically all ionic liquids. The preference for indole is especially large in polar ionic liquids, like [EMIM][Cl] or [BMIM][OAc], in which the anion offers ample opportunity for H-bonding, as is well known for the chloride and acetate anions.24 These same polar ionic liquids show hardly any preferential uptake of larger N-heterocycles like DMI or NMC in comparison with the S-heterocycles, and similar percentual uptakes are recorded for both compound classes (entries 1 to 3). These uptake values, typically below 10%, are particularly poor for the chloride ionic liquids.More interesting results are obtained with the bistriflimide (NTf2) ionic liquids. Irrespective of the cation, indole remains the preferred compound, but the uptake values for the other, larger N-heterocycles are 2 or 3 times higher than for the S-heterocycles. This indicates that this intrinsic selectivity is to be ascribed to an effect of the NTf2 anion, since EMIM, BMIM and even the non-aromatic BMPyrr ILs yield highly similar results (entries 4–6). In contrast with the Cl− and AcO− anions, the bistriflimide anion does not interact as strongly with the cation.25 This is reflected in the cation's increased ability to interact with the N-alkyl-substituted compounds like DMI and NMC, for instance by hydrogen bonding between the hydrogen atom at C2 in imidazolium rings and the substituted N in the heteroaromatic compound.26 The generally lower uptakes of heteroaromatics in the C2-substituted imidazolium IL [BDMIM][NTf2] (entry 8) when compared with [BMIM][NTf2] prove this hypothesis and show that polarity and/or the labile hydrogen on the imidazolium structure favour the interaction with the solutes. Increasing the cation polarity, e.g. by inserting a nitrile group in the side chain, generally raises the affinity for all heteroaromatic compounds, while the selectivity stays similar (entry 7) without indole but lower when taking into account this compound.
In order to fine-tune the properties of the bistriflimide ionic liquids, mixtures with ionic liquids containing other cations were employed. Generally, cations like choline or betainium seem to suppress thiophenes uptake in comparison with [BMIM][NTf2]; however this occurs slightly at the expense of the NMC and DMI uptake (entries 9 and 10). The more apolar methyltrioctylammonium increased again the affinity for the S-compounds (entry 11).
For its generally satisfactory performance, combining high uptake of nitrogen compounds and significant selectivity, [BMIM][NTf2] was chosen as a reference liquid phase for the rest of the N/S selective separations.
Admixture of metal ions to the ionic liquids
After the initial solvent selection, the same type of batch experiment was repeated but using [BMIM][NTf2] containing the bistriflimide salt of copper(I).† Copper(I) salts were chosen in view of the known affinity of Cu+-exchanged zeolites27 and Cu+-containing ionic liquids16 for heterocycles. In such cases, direct coordination of the heterocycle on the metal ion is invoked as one of the mechanisms leading to enhanced uptake or affinity. In order to map the effect of the copper compounds at various concentration levels, 0.04 M and 1 M solutions of CuNTf2 in [BMIM][NTf2] were prepared. During the biphasic experiments, this corresponds to initial (total heterocycle)/(metal ion) molar ratios of 1.5 and 0.06 respectively; the corresponding partial ratios (total N-heterocycle)/(metal ion) are 1 and 0.04. The results are shown in Fig. 1.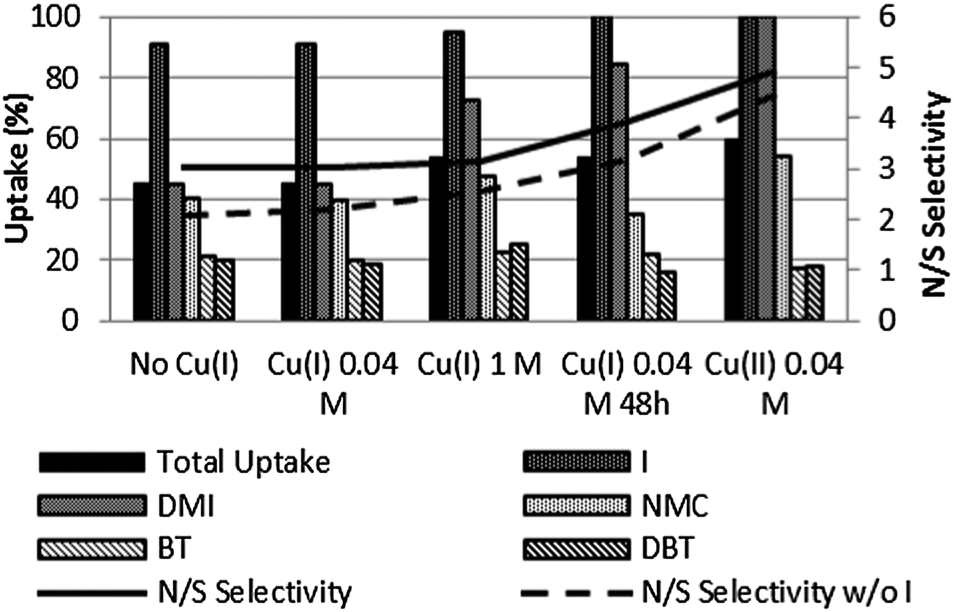 | ||
| Fig. 1 Comparison of the uptakes in the biphasic experiments using Cu-containing ionic liquid mixtures at 0.04 M and 1 M concentrations in [BMIM][NTf2]. The histograms show the percentual molar uptake of each impurity for each ionic liquid phase. The full lines indicates the N/S selectivities. Conditions as in Table 1. | ||
| Ionic liquidb | Uptake (molar%)c | N/S selectivityd | |||||||
|---|---|---|---|---|---|---|---|---|---|
| I | DMI | NMC | BT | DBT | Total | with I | without I | ||
|
a Batch experiment using 0.2 ml of ionic liquid and 1.7 ml of heptane/toluene model oil contaminated with 1.9 mM I, 1.5 mM DMI, 1.2 mM NMC, 1.6 mM BT and 1.2 mM DBT; equilibration for 0.75 h at RT.
b All IL mixtures are 1:1 molar ratios.
c Molar% of the compounds transferred to the IL phase; repetitions resulted in a fluctuation of less than 2%.
d N/S selectivity = (moles of N-heterocyclic compounds in the IL)/(moles of S-heterocyclic compounds in the IL). Values were calculated including indole or not.
e [C2NC1Pyrr] and [BDMIM] refer to the 1-(1-cyanomethyl)-1-methylpyrrolidinium and 1-butyl-2,3-dimethylimidazolium cations, respectively.
|
|||||||||
| 1 | [EMIM][Cl] | 96 | 3 | 4 | 5 | 2 | 28 | 11.5 | 1.2 |
| 2 | [BMIM][Cl | 96 | 6 | 7 | 6 | 4 | 29 | 8.0 | 1.3 |
| 3 | [BMIM][AcO] | 100 | 18 | 22 | 20 | 18 | 39 | 2.7 | 1.0 |
| 4 | [EMIM][NTf2] | 90 | 39 | 34 | 17 | 14 | 41 | 3.6 | 2.3 |
| 5 | [BMIM][NTf2] | 91 | 45 | 40 | 21 | 19 | 45 | 3.4 | 2.1 |
| 6 | [BMPyrr][NTf2] | 91 | 44 | 39 | 21 | 18 | 45 | 3.1 | 2.1 |
| 7 | [C2NC1Pyrr][NTf2]e | 93 | 47 | 49 | 24 | 25 | 49 | 2.6 | 2.0 |
| 8 | [BDMIM][NTf2]e | 81 | 28 | 12 | 7 | 2 | 30 | 8.7 | 4.3 |
| 9 | [BMIM][NTf2] + [Choline][NTf2] | 86 | 31 | 21 | 10 | 4 | 34 | 8.9 | 4.3 |
| 10 | [BMIM][NTf2] + [HBetaine][NTf2] | 93 | 43 | 19 | 7 | 3 | 39 | 9.6 | 6.0 |
| 11 | [BMIM][NTf2] + [N8881][NTf2] | 87 | 40 | 39 | 19 | 21 | 44 | 2.8 | 1.9 |
Upon increasing the concentration of the copper(I) compound in the IL from 0 to 1 M, there was some improvement in the overall sorption capacity and a somewhat increased affinity was noted for NMC and the sulfur compounds, when compared with previous experiments. However, the extent of the increase in copper concentration from 0.04 M to 1 M was, disappointingly, not reflected on the total heterocycle uptake, which only increased from 45% to 54%. This can be explained by the effect that the increase of the copper salt concentration has on the polarity of the IL phase, the increased polarity affecting the heterocyclic compounds' solubility in such a way that the specific interactions of the metal species with the heterocycles are not enough to stabilize the impurities in the IL phase. Surprisingly, when the mixture with the IL that contained 0.04 M copper(I) was left for a longer time (48 h), a trend towards a more selective N vs. S heterocycle uptake was observed. Although there was no change in the IL + Cu system's colour, this was attributed to the oxidation of the metal species to copper(II), in the presence of the model oil. Apparently, in the presence of the copper(II) compound, the ionic liquid phase increases its overall affinity for the N compounds, indole and dimethylindole in particular, while the thiophenes interact more poorly. Considering Pearson's HSAB concept of Lewis acidity/basicity, it must be noted that Cu2+ is a considerably harder Lewis acid than the very soft Cu+ ion, and therefore has less affinity for the softer Lewis bases in solution: the S heterocycles.28 This assumption was reinforced by the uptake and selectivity values obtained when performing the same experiment with an IL containing 0.04 M CuII(NTf2)2 (Fig. 1).† This IL phase presented a complete uptake of both I and DMI and an increased uptake of NMC, when compared with the copper(I)-containing IL phases, which is reflected in an increased N/S selectivity.
To assess in more detail the role of the nature of the metal ion, the same type of liquid–liquid extraction experiments was performed using ILs containing various metal salts, namely of copper(I), copper(II), zinc(II), iron(II) and iron(III) with bistriflimide or chloride anions.† All bistriflimide metal salts are in 0.04 M concentration for direct comparison with the previously investigated NTf2− salts of copper(I) and copper(II). Results are shown in Fig. 2. Chloride salts were prepared by mixing [BMIM][Cl] with the respective metal chlorides in a 1:1 or 1:2 metal/IL ratio required to form the respective [BMIM]y[MClx] ionic liquids, as shown in Fig. 3.29†
 | ||
| Fig. 2 Comparison of the uptakes in biphasic experiments using ILs containing different metal NTf2− salts. Conditions as in Table 1. | ||
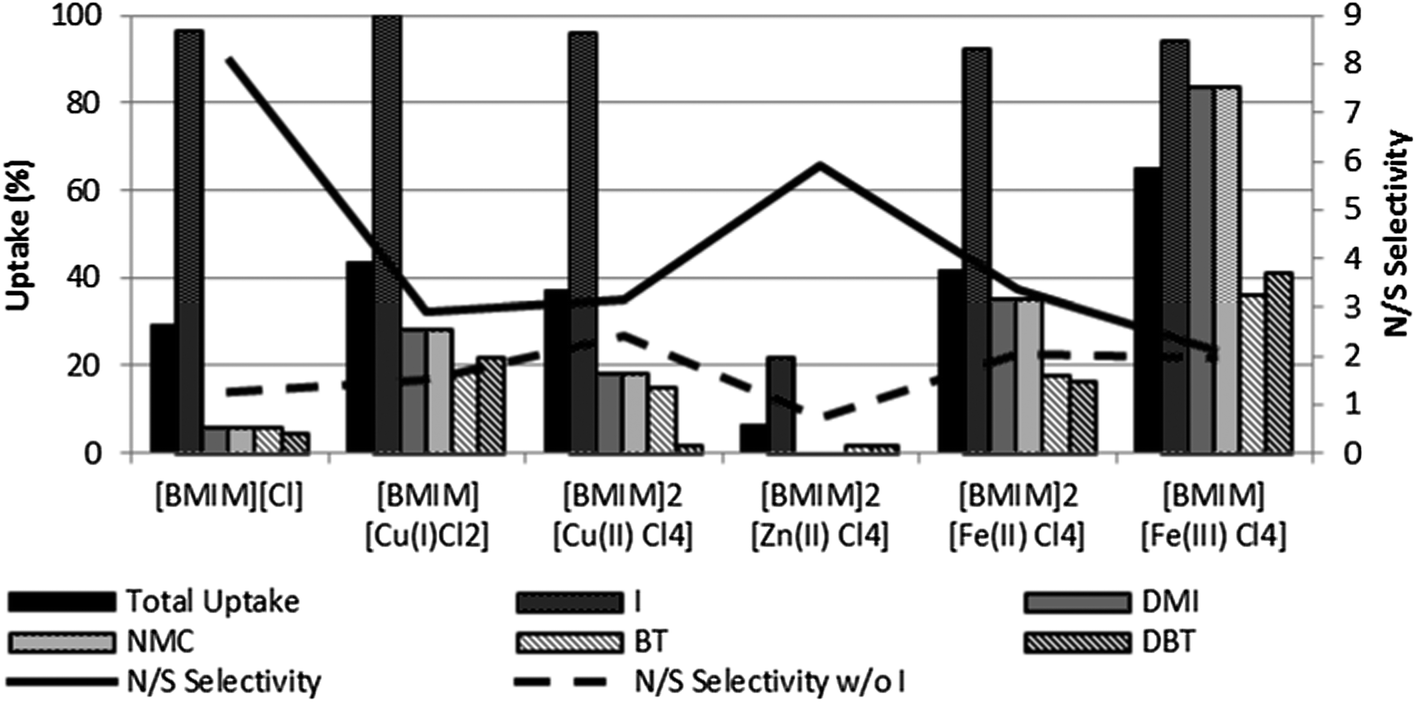 | ||
| Fig. 3 Comparison of the uptakes in biphasic experiments using ILs containing different metal chloride salts. Conditions as in Table 1. | ||
The bistriflimide salts offered, in general, a very good uptake of both I and DMI. In this group of ILs, CuII(NTf2)2 offered the best results in the uptake of all N-impurities in the model oil as well the best total heterocycle uptake. FeII(NTf2)2 afforded a similar total uptake but appeared to have significantly less affinity for the more apolar NMC. When comparing with the pure [BMIM][NTf2] IL, the advantage of the presence of the metal salt can clearly be seen: all the NTf2− metal salt-containing IL performed better than the metal-free [BMIM][NTf2], regarding both total uptake and selectivity.
When compared with their bistriflimide counterparts, the ILs containing the chloride salts of copper(I), copper(II) or iron(II) presented a low uptake of the more substituted N-compounds, DMI and NMC (Fig. 3). This resulted in a very poor N/S selectivity, especially when considering the values for DMI + NMC versus BT + DBT (“Selectivity w/o I”, in Fig. 3). This is in line with what was observed for the pure [BMIM][Cl] IL. However, the uptake of DMI and NMC was still larger than the one observed for the pure IL. The same trend for augmented uptake capacity in the presence of the metal species was also observed for the experiments with NTf2− compounds, as shown in Fig. 2. The [FeIIICl4]−-containing IL presented a total uptake capacity for the more apolar nitrogen compounds and even for the sulfur compounds which was larger than that of any of the other ILs described in this set and the previous, contrasting with the relatively low uptakes from the other IL + [MClx] systems. Noteworthy is the fact that, apart from [BMIM][FeIIICl4], all bistriflimide salt containing ILs offered a better total uptake and selectivity than their chloride salt counterparts even though the metal concentration was much lower.
Breakthrough experiments
After determining the most adequate metal/IL systems for selective denitrogenation, the next natural step, in view of a viable industrial application of such systems, is their immobilization on the pore surface of solid supports. The resulting supported ionic liquid phase (SILP) allows for application in breakthrough experiments. In these experiments, the ionic liquid was non-covalently adsorbed on the pore surface of a silica support which was inserted in a column. The model oil containing the N and S impurities was flushed through the column and the eluent was collected and analysed.Following the trends demonstrated in the liquid–liquid extraction experiments, breakthrough experiments were performed with SILP columns of [BMIM][NTf2] – either pure or loaded with 0.04 M CuII(NTf2)2 – and of [BMIM][Cl]/FeCl3.
Packed-bed columns
The packed-bed columns contained powdered mesoporous silica loaded with a SILP, and were used as stainless steel columns. The blank column containing only the solid silica support was also tested: all impurities eluted at the dead time of the column, implying no uptake occurred. Next, [BMIM][NTf2] was immobilized on the surface of the mesoporous silica in such an amount as to fill half of the solid's total pore volume (0.5 ml per g of mesoporous silica). The first run performed with this SILP (Fig. 4a) shows that the SILP causes a delay in the elution of indole but, unlike in the biphasic liquid–liquid experiments, none of the other N-compounds was significantly retained. Noticeably, the breakthrough profile of indole displays a broadened shape, which is indicative for too slow diffusion of the compound into the adsorbent phase.30 The column was regenerated with pure solvent mixture and used in a second run (Fig. 4b), this time using a model oil containing no indole, in an effort to determine whether the uptake of this compound impeded the uptake of the other compounds. Fig. 4b shows that the selectivity of the SILP for indole does not affect the affinity for the other N-compounds: again, neither DMI nor NMC were significantly retained.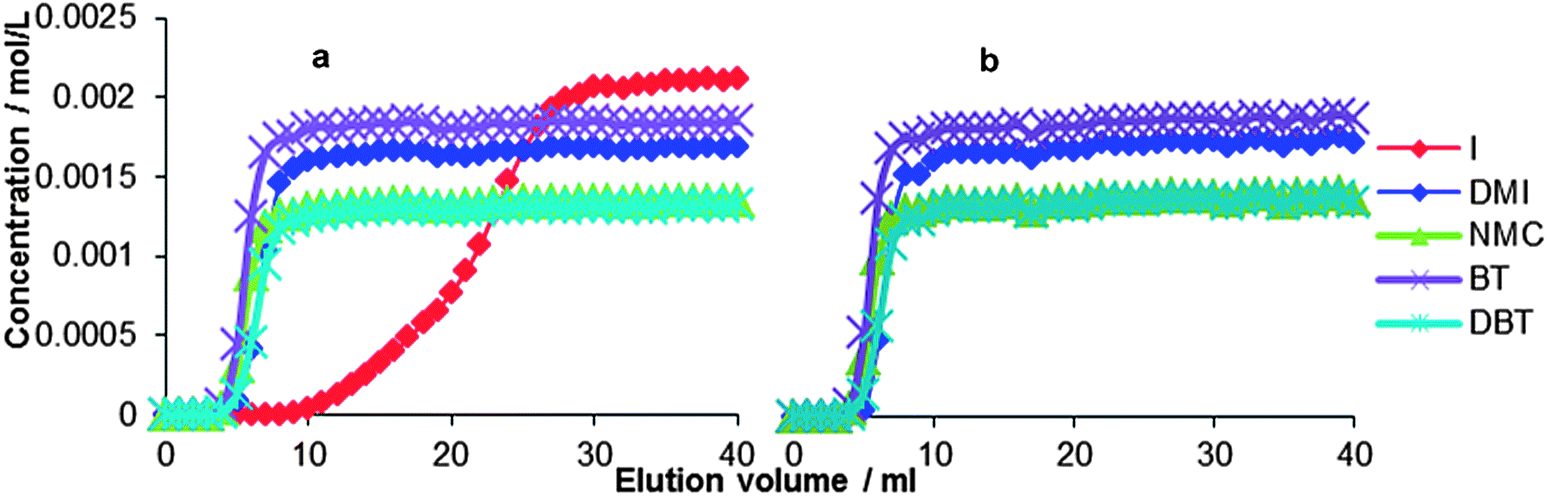 | ||
| Fig. 4 Breakthrough profiles, in 2 consecutive runs, for the pure [BMIM][NTf2] SILP packed-bed column at 1 ml min−1 flow rate: (a) experiment including indole; (b) experiment without indole. The column was abundantly rinsed with H/T 4:1 mixture in between the runs. A GC analysis of the eluent after the column regeneration showed no N or S impurity. | ||
In view of the good result obtained with the ionic liquid containing copper(II) in the liquid–liquid experiments (Fig. 1), two consecutive breakthrough experiments were run with a packed-bed column containing [BMIM][NTf2] with 4 × 10−2 M CuII(NTf2)2. Both runs presented near–identical profiles (Fig. 5a). Comparing these with the metal-free SILP profile (Fig. 4a), the copper(II)-containing SILP presents a similar profile but a sharper uptake of indole. This column displayed only a slight uptake of DMI, as seen in the first breakthrough profile of Fig. 5a. Given the discrepancy between the liquid–liquid and the breakthrough experiments in the uptake of the more apolar N-compounds (DMI and NMC), it was suspected that the contact times between the mobile phase and the SILP phase were not high enough for an efficient uptake. Hence, the copper(II)-SILP was also tested at a lower flow rate of 0.2 ml min−1 (Fig. 5b). In these conditions, the copper(II)-SILP packed-bed column presents a slightly better uptake of both DMI and NMC than at 1 ml min−1, but still the profiles did not match the expectations from the batch experiments.
 | ||
| Fig. 5 Breakthrough profiles with the CuII(NTf2)2 containing [BMIM][NTf2] SILP packed-bed column: (a) two consecutive runs at 1 ml min−1 flow rate, (b) run at 0.2 ml min−1 eluent flow rate. | ||
Monolithic silica columns
Next, similar experiments were performed, now using hierarchically structured silica monoliths as solid support. After a blank run, in which it was established that the monolith itself does not have a separation capacity, pure, metal-free [BMIM][NTf2] was immobilized onto the pore surface to create a SILP monolith column. Running the mobile phase at a flow rate of 0.2 ml min−1 afforded the profile on Fig. 6a. | ||
| Fig. 6 Breakthrough profiles with SILP containing monoliths: (a) SILP based on pure [BMIM][NTf2]; (b) and (c) two consecutive runs with a [BMIM][NTf2] SILP containing 4 × 10−2 M CuII(NTf2)2. Flow rate: 0.2 ml min−1. After each run, the column was regenerated by flushing with the pure solvent mixture. | ||
Comparison with the profile for the packed-bed columns at the same flow rate (Fig. 5b) shows that the SILP monolith presents the same adsorption profile, with the preferential uptake of indole. The column regeneration is also efficient, with the expected delay in the release of indole. A slight selectivity for the uptake of the apolar DMI and NMC is observed.
A new monolithic SILP column was then prepared with [BMIM][NTf2] containing 4 × 10−2 M CuII(NTf2)2 and the same experiment was run once again at 0.2 ml min−1 (Fig. 6b). The monolith column was regenerated and a second run was performed (Fig. 6c).
Comparing this result with the corresponding copper(II)-SILP packed-bed column at the same flow rate (Fig. 5b), it can be concluded that the monolith plays a decisive role in the uptake process. The hierarchical structure of the monolith, with macro- and mesopores, aids the diffusion of the solutes and increases the SILP's contact with the bulky N- and S-compounds. As a result, the preferences of the SILP monolithic system perfectly mirror those of the same ionic liquid in batch mode (Fig. 2), with a decreasing preference in the order: I > DMI, NMC > BT, DBT. Importantly, the nitrogen contaminants are well separated from the sulfur contaminants. The similar breakthrough profiles obtained for both runs show that this system can be successfully regenerated and used in multiple cycles. Moreover, the identical profiles indicate that no significant leaching of the ionic liquid phase nor any permanent chemical modification of the metal-containing SILP during the process takes place.
The good performance of the monolithic silica supports, and the very good result obtained with the iron(III) tetrachloride IL in the liquid–liquid experiments (Fig. 3) motivated the application of this IL in breakthrough runs using a monolith column. As seen in Fig. 7, indole is taken up until about 20 ml into the experiment, in the same manner as on the metal-free SILP monolith column (Fig. 6a). However, in this case, 1,2-dimethylindole is also taken up in the same extent, resulting in almost 20 ml of model oil containing only 33% of its normal nitrogen content. Comparing with the corresponding liquid–liquid experiment performed with the same IL (Fig. 3), there is now a more marked selectivity towards the nitrogen compounds. While NMC was also efficiently retained in the Cu-doped [BMIM][NTf2] monolith, this compound is less efficiently withheld on the FeIIICl4−-containing ionic liquid. This is somehow at contrast with the liquid–liquid equilibrium data of Fig. 3, and suggests that, in the breakthrough test with the FeIIICl4−-containing ionic liquid, adsorption equilibrium was not reached for NMC due to the slow diffusion of this large compound.
 | ||
| Fig. 7 Breakthrough profile at 0.2 ml min−1 of the Fe(III)Cl4-SILP monolith column. | ||
Experimental section
Bistriflimide ionic liquids and [BMIM][Cl] were obtained commercially with a purity of ≥95%. The ILs with anions of the type [MClx] were prepared by mixing [BMIM][Cl] and the metal chloride in the appropriate proportions at room temperature.29 All syntheses of metal bistriflimide salts and ILs of the type [BMIM]y[MClx] are described in the ESI.† Residual solvent and water were removed under vacuum after the formation of the IL. All ILs were dried at <0.1 mbar vacuum at 90 °C overnight prior to use.The model oil was prepared by dissolving 55 mg of each compound (indole, 1,2-dimethylindole, N-methylcarbazole, benzothiophene and dibenzothiophene) in 250 ml of a solvent mixture composed of heptane and toluene in a 4:1 volume ratio.
General procedure for the biphasic liquid–liquid extraction experiments
The experiments were performed, in triplicate when possible, in GC vials with 0.2 ml of IL phase and the rest of the vials' volume (1.7 ml) containing the model oil with the N- and S-compounds, leaving no headspace. The mixtures were stirred for 45 minutes at room temperature, after which period the model oil phase was analysed with GC. Each liquid–liquid experiment was accompanied by a blank in which the vial was filled only with the model oil and no IL phase. No solvent uptake was observed.General procedure for SILP powder preparation
Silica gel, large pore from Alfa Aesar (58 μm particle size, 300 m2 g−1 surface area, 1.6 ml g−1 pore volume, 15 nm pore diameter) was used. The appropriate volume of dry ionic liquid to prepare a SILP with half of the solid's initial pore volume filled was dissolved in 50 ml of anhydrous dichloromethane. This solution was added to the silica, previously activated for 3 h at 500 °C and cooled under vacuum, and the mixture was stirred for 2 h under N2 atmosphere. The solvent was removed under vacuum, resulting in a white, free-flowing powder. The SILP powder was packed into the 10 cm long, 4 mm inner diameter stainless steel column (1.3 ml inner cylindrical volume) under N2 atmosphere.General procedure for SILP monolith preparation
This type of column consists of a cylindrical silica monolith encased between two glass tubes and in a Teflon sheath. The monoliths used have a macropore diameter of 5–6 μm, mesopore diameter of 11 nm, a specific surface area of 550 m2 g−1, mesopore volume 1.1 ml g−1 and macropore volume of 1.7 ml g−1. They have a diameter of 6 mm and 4 cm in length, corresponding to a cylindrical volume of 1.1 ml. The synthesis of the monoliths is described in the literature.21b The dry ionic liquid was dissolved in an amount of dry dichloromethane such as that the resulting solution's volume was the same as the monolith's total pore volume. The SILP was prepared by filling the entire pore volume of the monolith with the solution. The amount of IL was such as to fill half the monolith's total pore volume, as in the preparation of the powder SILPs. The solvent was removed and the SILP-containing monolith dried overnight under vacuum. This SILP column was fitted to the HPLC pump directly by the glass ends.General procedure for the breakthrough experiments
A typical breakthrough experiment was performed by attaching a stainless steel packed-bed column or a glass-ended, Teflon-sheathed monolith column to a HPLC pump. The column was then conditioned with at least 20 ml of pure solvent mixture at 1 ml min−1 (0.5 ml min−1 for the monoliths). The model oil was eluted at the chosen flow rate, at which point t = 0. Each millilitre of the eluent was collected and analysed with GC. The dead volume for a normal system, was approximately 4 ml. Blank experiments were performed by using a column containing only the respective solid support without any IL phase and eluting the model oil. The blank breakthrough profiles show no preferential uptake of any compound.Conclusions
The results shown allow the conclusion that ionic liquids containing the bistriflimide and chloride salts of copper(II) and iron(III), respectively, show a promising selectivity for the uptake of heteroaromatic nitrogen compounds when in the presence of other impurities usually found in fuel feeds, namely structurally similar heteroaromatic sulfur compounds. The ionic liquid [BMIM][NTf2] containing CuII(NTf2)2 was found to perform better than its copper(I) counterpart, affording a higher uptake and better selectivity in the uptake of N compounds over S. This species' harder Lewis acid character accounts for lower affinity for the softer sulfur compounds. Ionic liquids containing metal bistriflimide salts in a relatively low concentration were shown to outperform the current literature staple for metal-containing, IL-based extractive denitrogenation, [BMIM]y[MXx] ionic liquids,11d,e,h,14,16 in which the metal halide (MXx−n) is present in a very high 1:1 or 1:2 proportion. Metal bistriflimide IL systems present, therefore, a more benign alternative to the use of corrosive metal halides for the purification of fuels feeds.
The best performing IL systems were non-covalently immobilized on the pore surface of hierarchically structured silica monoliths to form SILP columns. This allowed the run of breakthrough experiments using these columns. The breakthrough profiles of the impurities showed that these monolithic SILPs are efficient materials for the selective denitrogenation of fuel feeds. A complete separation between N and S impurities was obtained when using a SILP consisting of a CuII(NTf2)2-containing [BMIM][NTf2] ionic liquid. The monolithic supports were shown to be determinant parameters in improving the performance of the SILPs. It is proposed that this derives from their hierarchical structure. The presence of the macropore/mesopore system optimizes the mass transport while the higher practical specific surface means that there is a higher SILP surface available to interact with the eluent, increasing their efficiency. Furthermore, it is shown that these heterogeneous systems are stable in the conditions used and can be applied in successive runs to denitrogenate fuel feeds.
Acknowledgements
This work was supported by 7PCRD EU funds from the Marie-Curie initial Training Network NANO-HOST (grant agreement ITN 215193). DDV is grateful to KULeuven and Belspo for Metusalem CASAS and IAP 7/05 grants respectively.Notes and references
- (a) A. Stanislaus, A. Marafi and M. S. Rana, Catal. Today, 2010, 153, 1–68 CrossRef CAS PubMed; (b) P. S. Kulkarni and C. A. M. Afonso, Green Chem., 2010, 12, 1139–1149 RSC.
- (a) M. Breysse, G. Djega-Mariadassou, S. Pessayre, C. Geantet, M. Vrinat, G. Perot and M. Lemaire, Catal. Today, 2003, 84, 129–138 CrossRef CAS; (b) L. Li, Y. Zhu, X. Lu, M. Wei, W. Zhuang, Z. Yang and X. Feng, Chem. Commun., 2012, 48, 11525–11527 RSC.
- (a) I. V. Babich and J. A. Moulijn, Fuel, 2003, 82, 607–631 CrossRef CAS; (b) M. J. Girgis and B. C. Gates, Ind. Eng. Chem. Res., 1991, 30, 2021–2058 CrossRef CAS.
- (a) M. Te, C. Fairbridge and Z. Ring, Appl. Catal., A, 2001, 219, 267–280 CrossRef CAS; (b) Y. Shiraishi, K. Tachibana, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 2002, 41, 4362–4375 CrossRef CAS.
- (a) L. C. Caero, J. F. Navarro and A. Gutierrez-Alejandre, Catal. Today, 2006, 116, 562–568 CrossRef PubMed; (b) Z. E. A. Abdalla and B. Li, Chem. Eng. J, 2012, 200, 113–121 CrossRef PubMed; (c) P. Yin, J. Wang, Z. Xiao, P. Wu, Y. Wei and T. Liu, Chem.–Eur. J., 2012, 18, 9174–9178 CrossRef CAS PubMed.
- L. Y. Kong, G. Li and X. S. Wang, Catal. Lett., 2004, 92, 163–167 CrossRef CAS.
- X. L. Ma, A. N. Zhou and C. S. Song, Catal. Today, 2007, 123, 276–284 CrossRef CAS PubMed.
- (a) J. M. Campos-Martin, M. C. Capel-Sanchez, P. Perez-Presas and J. L. G. Fierro, J. Chem. Technol. Biotechnol., 2010, 85, 879–890 CrossRef CAS; (b) Z. Ismagilov, S. Yashnik, M. Kerzhentsev, V. Parmon, A. Bourane, F. M. Al-Shahrani, A. A. Hajji and O. R. Koseoglu, Catal. Rev., 2011, 53, 199–255 CrossRef CAS.
- (a) Y. H. Jia, G. Li, G. L. Ning and C. Z. Jin, Catal. Today, 2009, 140, 192–196 CrossRef CAS PubMed; (b) M. Macaud, M. Sévignon, A. Favre-Réguillon, M. Lemaire, E. Schulz and M. Vrinat, Ind. Eng. Chem. Res., 2004, 43, 7843–7849 CrossRef CAS; (c) H. Yang, J. Chen, C. Fairbridge, Y. Briker, Y. J. Zhu and Z. Ring, Fuel Process. Technol., 2004, 85, 1415–1429 CrossRef CAS PubMed.
- M. Francisco, A. Arce and A. Soto, Fluid Phase Equilib., 2010, 294, 39–48 CrossRef CAS PubMed.
- (a) L. Alonso, A. Arce, M. Francisco and A. Soto, J. Chem. Eng. Data, 2010, 55, 3262–3267 CrossRef CAS; (b) A. Bosmann, L. Datsevich, A. Jess, A. Lauter, C. Schmitz and P. Wasserscheid, Chem. Commun., 2001, 2494–2495 RSC; (c) J. Esser, P. Wasserscheid and A. Jess, Green Chem., 2004, 6, 316–322 RSC; (d) E. S. Huh, A. Zazybin, J. Palgunadi, S. Ahn, J. Hong, H. S. Kim, M. Cheong and B. S. Ahn, Energy Fuels, 2009, 23, 3032–3038 CrossRef CAS; (e) N. H. Ko, J. S. Lee, E. S. Huh, H. Lee, K. D. Jung, H. S. Kim and M. Cheong, Energy Fuels, 2008, 22, 1687–1690 CrossRef CAS; (f) M. Matsumoto, M. Mikami and K. Kondo, J. Jpn. Pet. Inst., 2006, 49, 256–261 CrossRef CAS; (g) L. L. Xie, X. Chen, X. X. Wang, X. Z. Fu, A. Favre-Reguillon, S. Pellet-Rostaing and M. Lemaire, Chin. J. Inorg. Chem., 2008, 24, 919–925 CAS; (h) S. G. Zhang, Q. L. Zhang and Z. C. Zhang, Ind. Eng. Chem. Res., 2004, 43, 614–622 CrossRef CAS.
- (a) V. I. Parvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef CAS PubMed; (b) H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef CAS PubMed; (c) Q. H. Zhang, S. G. Zhang and Y. Q. Deng, Green Chem., 2011, 13, 2619–2637 RSC.
- (a) D. S. Zhao, J. L. Wang and E. P. Zhou, Green Chem., 2007, 9, 1219–1222 RSC; (b) H. M. Li, W. S. A. Zhu, Y. Wang, J. T. Zhang, J. D. Lu and Y. S. Yan, Green Chem., 2009, 11, 810–815 RSC; (c) Y. Chao, H. Li, W. Zhu, G. Zhu and Y. Yan, Pet. Sci. Technol., 2010, 28, 1242–1249 CrossRef CAS; (d) X. C. Chen, D. D. Song, C. Asumana and G. R. Yu, J. Mol. Catal. A: Chem., 2012, 359, 8–13 CrossRef CAS PubMed; (e) C. Zhang, X. Y. Pan, F. Wang and X. Q. Liu, Fuel, 2012, 102, 580–584 CrossRef CAS PubMed; (f) D. Liu, J. Gui, D. Liu, X. Peng, S. Yang and Z. Sun, Energy Sources, Part A, 2013, 35, 1–8 CrossRef CAS; (g) Y. Nie, Y. X. Dong, L. Bai, H. F. Dong and X. P. Zhang, Fuel, 2013, 103, 997–1002 CrossRef CAS PubMed; (h) W. S. Zhu, H. M. Li, X. Hang, Y. S. Yan, H. D. Lu and J. X. Xia, Energy Fuels, 2007, 21, 2514–2516 CrossRef CAS.
- N. V. Likhanova, D. Guzman-Lucero, E. A. Flores, P. Garcia, M. A. Dominguez-Aguilar, J. Palomeque and R. Martinez-Palou, Mol. Diversity, 2010, 14, 777–787 CrossRef CAS PubMed.
- E. Kuhlmann, M. Haumann, A. Jess, A. Seeberger and P. Wasserscheid, ChemSusChem, 2009, 2, 969–977 CrossRef CAS PubMed.
- C. P. Huang, B. H. Chen, J. Zhang, Z. C. Liu and Y. X. Li, Energy Fuels, 2004, 18, 1862–1864 CrossRef CAS.
- (a) A. Riisager, R. Fehrmann, M. Haumann and P. Wasserscheid, Top. Catal., 2006, 40, 91–102 CrossRef CAS PubMed; (b) C. Van Doorslaer, J. Wahlen, P. Mertens, K. Binnemans and D. De Vos, J. Chem. Soc., Dalton Trans., 2010, 39, 8377–8390 RSC; (c) C. F. Poole and S. K. Poole, J. Chromatogr., A, 2010, 1217, 2268–2286 CrossRef CAS PubMed.
- (a) C. P. Mehnert, R. A. Cook, N. C. Dispenziere and M. Afeworki, J. Am. Chem. Soc., 2002, 124, 12932–12933 CrossRef CAS PubMed; (b) C. P. Mehnert, Chem.–Eur. J., 2005, 11, 50–56 CrossRef PubMed.
- C. G. Frost and L. Mutton, Green Chem., 2010, 12, 1687–1703 RSC.
- A. Stankiewicz, Chem. Eng. Sci., 2001, 56, 359–364 CrossRef CAS.
- (a) A. El Kadib, R. Chimenton, A. Sachse, F. Fajula, A. Galarneau and B. Coq, Angew. Chem., Int. Ed., 2009, 48, 4969–4972 CrossRef CAS PubMed; (b) A. Sachse, A. Galarneau, F. Fajula, F. Di Renzo, P. Creux and B. Coq, Microporous Mesoporous Mater., 2011, 140, 58–68 CrossRef CAS PubMed.
- A. Sachse, A. Merceille, Y. Barre, A. Grandjean, F. Fajula and A. Galarneau, Microporous Mesoporous Mater., 2012, 164, 251–258 CrossRef CAS PubMed.
- (a) J. J. Rodriguez, J. Lemus, J. Palomar and M. A. Gilarranz, Adsorption, 2011, 17, 561–571 CrossRef; (b) A. Chrobok, S. Baj, W. Pudlo and A. Jarzebski, Appl. Catal., A, 2009, 366, 22–28 CrossRef CAS PubMed; (c) A. Chrobok, S. Baj, W. Pudlo and A. Jarzebski, Appl. Catal., A, 2010, 389, 179–185 CrossRef CAS PubMed.
- S. Chen, R. Vijayaraghavan, D. R. MacFarlane and E. I. Izgorodina, J. Phys. Chem. B, 2013, 117, 3186–3197 CrossRef CAS PubMed.
- A. Kobayashi, K. Osawa, M. Terazima and Y. Kimura, Phys. Chem. Chem. Phys., 2012, 14, 13676–13683 RSC.
- C. C. Cassol, A. P. Umpierre, G. Ebeling, B. Ferrera, S. S. X. Chiaro and J. Dupont, Int. J. Mol. Sci., 2007, 8, 593–605 CrossRef CAS.
- (a) A. J. Hernandez-Maldonado and R. T. Yang, Angew. Chem., Int. Ed., 2004, 43, 1004–1006 CrossRef CAS PubMed; (b) A. J. Hernandez-Maldonado, F. H. Yang, G. S. Qi and R. T. Yang, J. Chin. Inst. Chem. Eng., 2006, 37, 9–16 CAS; (c) A. Jayaraman, F. H. Yang and R. T. Yang, Energy Fuels, 2006, 20, 909–914 CrossRef CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- (a) S. A. Bolkan and J. T. Yoke, J. Chem. Eng. Data, 1986, 31, 194–197 CrossRef CAS; (b) T. Sasaki, C. M. Zhong, M. Tada and Y. Iwasawa, Chem. Commun., 2005, 2506–2508 RSC; (c) K. R. Seddon, J. Estager, P. Nockemann, M. Swadzba-Kwasny and S. Tyrrell, Inorg. Chem., 2011, 50, 5258–5271 CrossRef PubMed; (d) M. S. Sitze, E. R. Schreiter, E. V. Patterson and R. G. Freeman, Inorg. Chem., 2001, 40, 2298–2304 CrossRef CAS PubMed.
- E. Jolimaitre, K. Ragil, M. Tayakout-Fayolle and C. Jallut, AIChE J., 2002, 48, 1927–1937 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed descriptions of the syntheses of the bistriflimide metal salts and [BMIM]y[MClx] ILs. See DOI: 10.1039/c3ra43585g |
| This journal is © The Royal Society of Chemistry 2014 |