
Enantioselective separation of chiral ofloxacin using functional Cu(ii)-coordinated G-rich oligonucleotides†
Abstract
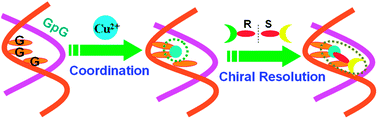
The DNA-based selector for discriminating chiral ofloxacin with high enantioselectivity and affinity is constructed through Cu(II)-coordination with G-rich duplex containing successive guanines. Using this chiral selector, R- and S-ofloxacin can be directly enriched from the racemate, with the enantiomeric excess of 85% (R) and 78% (S) individually by three operational stages.
 Please wait while we load your content...
Please wait while we load your content...

