
Density functional calculations on the distribution of Ti in a Y zeolite and its influence on acidity
Abstract
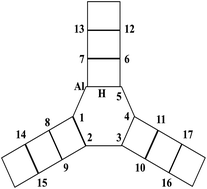
The question of whether Ti replaces Al or Si in the framework of a Y zeolite is studied using a density function theory (DFT) calculation. The distribution properties of Ti in a zeolite are discussed by comparing substitution energies, they show that of the seventeen Si positions investigated the preferred substitution is the one where the Si atoms are located next to the Al, and that the Al is more easily replaced than Si. The framework of the Y zeolite shows a local expansion and deformation after the substitution. The deprotonation energy then reflects the change in Brönsted acidity, demonstrating that the replacement enhances the Brönsted acidity of the zeolite. Further, a detailed illustration of the relationship between the acidity of the zeolite and the location of the Ti is presented.
 Please wait while we load your content...
Please wait while we load your content...

