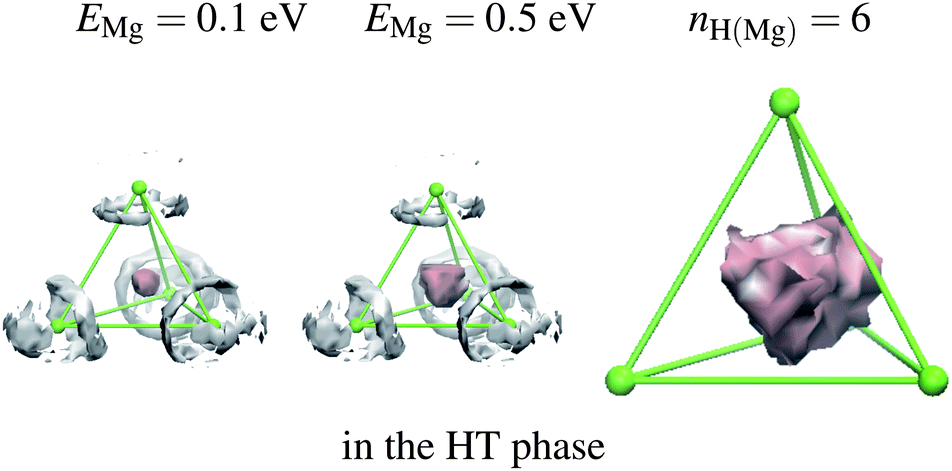DOI:
10.1039/C3RA42453G
(Paper)
RSC Adv., 2014,
4, 1366-1370
Magnesium ion dynamics in Mg(BH4)2(1−x)X2x (X = Cl or AlH4) from first-principles molecular dynamics simulations
Received
17th May 2013
, Accepted 11th November 2013
First published on
12th November 2013
Abstract
We use large-scale first-principles molecular dynamics simulations to find possible ways to make magnesium dication conductors based on Mg(BH4)2. The Mg atoms are confined in a tetrahedral cage of BH4 groups and coordinated by eight hydrogen atoms from these groups. These Mg atoms do not move outside the cage either in high- or low-temperature phases. After introducing larger-sized AlH−4 anions by substitution of 20% BH−4, the Mg atoms are able to move outside the cage with coordination from hydrogen atoms of BH−4 located outside the cage. Substitution by Cl− and I− anions did not allow large Mg movements.
1 Introduction
Ions in solid electrolytes generally do not move easily. These have very low diffusion coefficients except for some electrolytes. In these fast ion conductors, many consist of monovalent ions. Because of strong Coulombic interactions with counter ions, divalent ions have low ion conductivity; an exception is O2− conduction in Y-stabilized ZrO2. Higher valence positive-ions, because of their small size, are strongly trapped by the counter ions, resulting in a low ionic conductivity. If able to be developed, a fast dication solid conductor would add impetus in attaining high capacity rechargeable batteries with no liquid in the form of a “Mg|Mg2+ solid electrolyte|oxidant”. High ionic conductivities reported for Mg are limited to 10−4 S cm−1 at ∼800 K (ref. 1 and 2) and 10−6 S cm−1 at ∼430 K.3
We found experimentally4 fast ionic conduction of Li in the high-temperature (HT) phase of LiBH4 and uncovered its mechanism using large-scale first-principles molecular dynamics simulations.5,6 A Li atom moves to a metastable site, and then, a neighboring site Li atom moves to the vacancy generated. BH4 is almost in free rotation in the HT phase (P63mc), but is fixed in the low-temperature (LT) phase (Fddd), which has low Li ion conductivity. The former has higher symmetry than the latter, because BH4 looks more spherical at higher temperatures. If Li atom moves, BH4 rotates to maintain a high H-coordination number, resulting in a stable configuration while passing the activation state.7 Therefore, free rotation of BH4 promotes ionic conduction. A straightforwardly expected material for Mg ion conductor is Mg(BH4)2. Various structures have been proposed experimentally and calculated using density functional theory (DFT) calculations.8–14 These structures have only small differences in free energy.15 We have chosen HT and LT phases having large unit cells, with hexagonal (P61)16,17 and orthorhombic (Fddd)16 lattices, respectively. Mg(BH4)2 itself could have low ion conductivity either in the HT or LT phase. With the analogy of anion substitution in LiBH4 to increase the conductivity in the HT phase,18,19 we begin with the dynamics of the Mg atoms in Mg(BH4) using first-principles molecular dynamics simulations and then examine the effect of substitution by Cl−, I−, and AlH−4 anions for BH−4. Finally, a possible mechanism to increase conductivity is discussed. We have recently presented a short report on the effect of AlH4 substitution for BH4 in the HT phase of Mg(BH4)2.20 In this article, we extend the study to the LT phase and other anions to find a general way to increase the Mg dication conductivity.
2 Calculations
In the majority of the FPMD simulations, the electronic structure of the system was calculated based on DFT21,22 using the local density approximation, norm-conserving pseudopotentials,23,24 and Γ-point sampling of the Brillouin zone. The orbitals were expanded by the adaptive finite element basis functions as implemented in our DFT code “FEMTECK”,25,26 which is specially designed to perform fast FPMD for systems of light elements. Average cutoff energies of 70–80 Ry were used, while the resolution was enhanced near the atoms by adaptation of the grids.27 The equations of motion were integrated using the velocity-Verlet algorithm with a time step of 0.73 fs. The electronic states were quenched to the Born–Oppenheimer surface at each MD step using the limited-memory variant of the quasi-Newton method.28,29 For structure optimization and FPMD including iodine atoms, commercially available code, VASP (Vienna Ab initio Simulation Package),30,31 was used in which plane waves for orbitals, PAW pseudopotentials for atomic inner cores, PBE exchange functional, cutoff energy of 400 eV for orbitals and 3200 eV for electron density, and k-point sampling of the Γ point only in the FPMD calculations and 4 4 4 points (for Mg(AlH4)2) or 4 2 1 points (for the other systems) in structure optimizations were used.
The LT phase Mg(BH4)2was modeled using 660 atoms in a supercell of size 10.32 × 17.87 × 37.00 Å3, which is double the primitive unit cell.17 The HT phase was modeled using 704 atoms in a 10.65 × 18.83 × 36.90 Å3 cell the same size as the primitive unit cell. In simulations using FEMTECK the total simulation time was 10–15 ps. A snapshot of the FPMD simulation for the HT phase is shown in Fig. 1. The simulation temperature was controlled using the Berendsen thermostat with target temperatures of 450 K and 500 K for the LT and HT phases, respectively. Atomic masses were optimized following ref. 32 FPMD by VASP was performed in the same way using the same parameters as those in FEMTECK, but the total simulation time was 1.5 ps. Structure optimization was performed for the same models. For structure optimization of Mg(AlH4)2, an 8-fold super cell in P3m133 was used.
 |
| | Fig. 1 A snapshot of FPMD simulation of Mg(BH4)1.6 (AlH4)0.4 in the HT phase. Two tetrahedral cages sharing an apex are indicated by red lines. Green ball, B; large blue ball, Al; pink ball, Mg; small white ball, H. | |
3 Results and discussion
3.1 Pure Mg(BH4)2
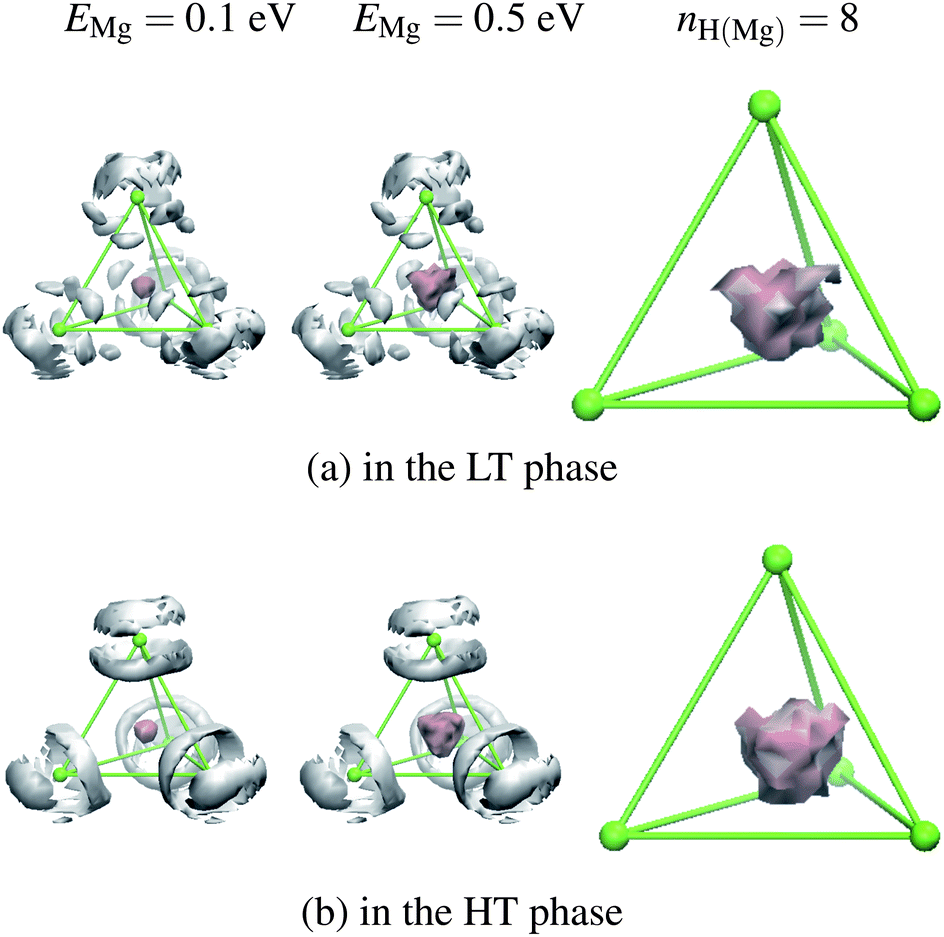
All Mg ions in either HT or LT phases are located in several kinds of tetrahedral cages composed of B atoms of BH4 as depicted in Fig. 1. Each cage is connected by sharing a B atom at an apex. To achieve Mg ion conduction, Mg atoms need to move outside of their tetrahedral cages. To see how Mg atoms are moving inside the cage, we calculated the Mg-atom distribution from the FPMD trajectories as shown in Fig. 2, in which the H-atom distribution is also shown. Although tetrahedral cages are dynamically distorted, all sides are normalized to unity in these figures by expressing the position q of the Mg atom with three vectors, ei (i = 1, 2, 3) from an apex of the tetrahedron to the other three apices,| | | q = q1e1 + q2e2 + q3e3. | (1) |
 |
| | Fig. 2 Time average Mg (pink regions at the center) and H (white regions at the corner) atom distributions in the tetrahedral cage (green bars) in Mg(BH4)2. Isosurfaces of potential EMg and H-coordination number nH(Mg) of Mg are shown. The H-distribution is at EH = 0.05 eV. A boron atom (green ball) is located at each apex of the tetrahedron. The solid surface shows the H-coordination only with the H atoms of BH4 at the apices of the tetrahedron. The transparent cloud (pale pink) includes the H atoms of the BH4 from the neighboring tetrahedral cage. | |
A set of (q1, q2, q3) is then used to express the projected position q′ as
| | | q′ = q1e′1 + q2e′2 + q3e′3, | (2) |
where the
e′i are three vectors directed towards the apices of the regular tetrahedron with unit sides. The atom distribution is given as an isopotential surface,
E, which is calculated from the Boltzmann distribution of atomic density
ρ,
E =
kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
ln(
ρ/
ρ0), where
kB and
T are Boltzmann constant and temperature, respectively. The origin of the potential is the most stable site,
i.e. at the center of the cage, where
ρ =
ρ0. From these distributions, the Mg atoms are found to be always within the tetrahedral cage, and not moving outside of the cage.
3.2 Coordination numbers
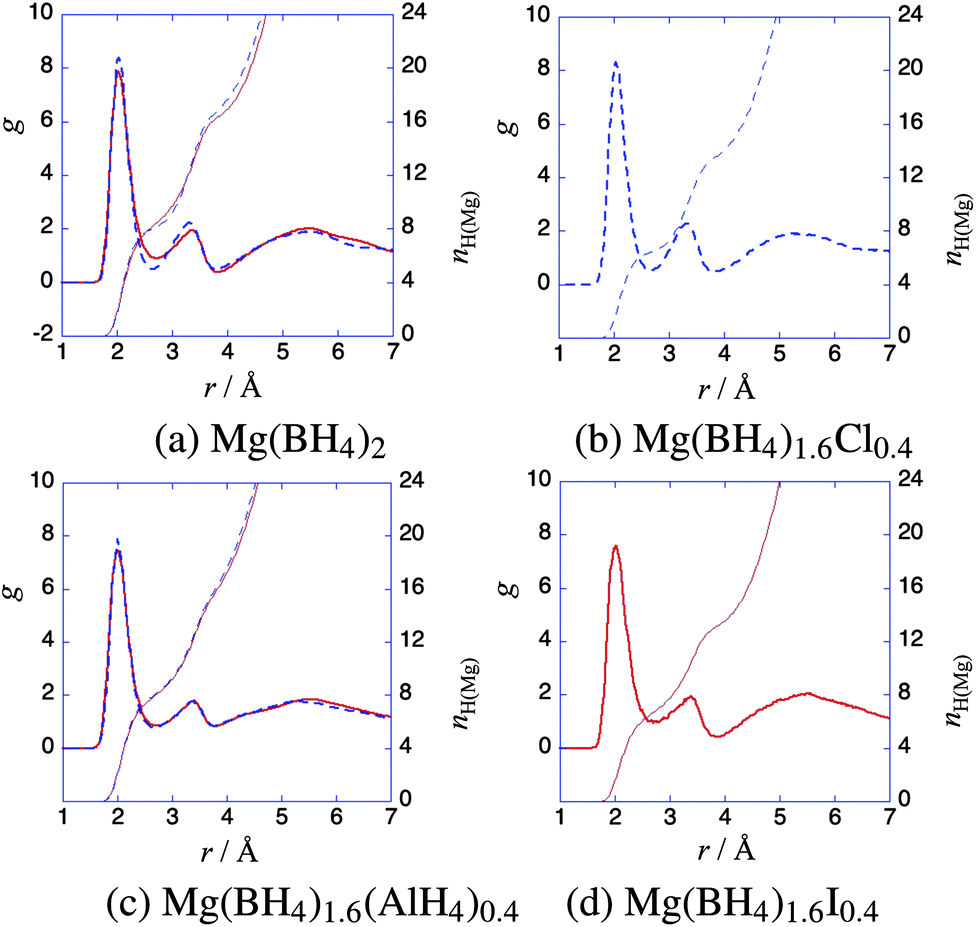
Because the charge on H is −(0.60–0.62)e,10 the Mg atoms are coordinated with H atoms. The H-coordination number of the Mg atoms is about 8 both in the HT and LT phases, as can be seen from radial distribution function (RDF, g) and its integration (=H-coordination number, nH(Mg)) in Fig. 3(a). The same number for the H-coordination was reported from neutron diffraction measurements.8 Because a B atom is located at an apex shared by two tetrahedrons, half of the four H atoms in BH4 are used to coordinate with a Mg atom in the cage and the other two H atoms are with a Mg atom of a neighboring cage. The H-distribution (Fig. 2) confirms the nearly free rotation of BH4 around the perpendicular lines from the apex in the HT phase. In the LT phase, H atoms are more restricted in their positions.
 |
| | Fig. 3 RDF (g) between Mg and H atoms and H-coordination number (nH(Mg)) of Mg. Red solid lines, HT phase; blue broken lines, LT phase; thick lines, g; thin lines, nH(Mg). | |
3.3 Substituted Mg(BH4)2
To see the effect of substitution, we have replaced 20% of the BH4 groups with the smaller ion Cl (radius = 1.81 Å) and with the larger ion AlH4 (Al–H distance = 1.6 Å). To ascertain the role of H atom in the atomic movement, a larger ion I− was also tested. The smaller ion is expected to trap cations inside of the cage whereas the larger ion assists movement of Mg outside because of a weaker trap. Substitution by the smaller ion Cl for BH4 does not change the Mg and H-distributions, as shown in Fig. 3(b) and 4. B–Cl and B–B distances are both 3.8 Å. For substitution by AlH4, a large influence is observed as shown in Fig. 5. The RDF becomes smoother (Fig. 3(c)). Even though four sides of the tetrahedron are different in length, all sides are normalized to unity as described by eqn (2). To avoid a numerical problem (division by a small value in the transformation of the coordinate system), the calculation of the distribution is skipped if the volume of the tetrahedron becomes too small (<7 Å3). From the figure, Mg ions now move to the outside of the cage, but the overall diffusion of Mg was not observed within a simulation time of 10 ps. This large displacement within the cage will be experimentally observed as a peak broadening in the X-ray diffraction determined by the atomic temperature factor B,34where q0 is the equilibrium position of an atom. From the listing of these values for Mg atom in Table 1, AlH4 substitution has a significant effect on the large Mg ion displacement.
 |
| | Fig. 4 Mg- and H-atom distributions in the tetrahedral cage of the LT phase Mg(BH4)1.6(Cl)0.4. Different anions, BH4 or Cl, are randomly placed at each apex of the tetrahedron. Isosurface of nH(Mg) = 6 is shown. See note in Fig. 2. | |
 |
| | Fig. 5 Mg- and H-atom distributions in the tetrahedral cage in Mg(BH4)1.6(AlH4)0.4. Different anions, BH4 or AlH4, are randomly placed at each apex of the tetrahedron. See note in Fig. 2. | |
Table 1 Atomic temperature factor B (in Å2) of Mg atoms
| System |
LT |
HT |
| Mg(BH4)2 |
1.6 |
1.9 |
| Mg(BH4)1.6Cl0.4 |
1.7 |
|
| Mg(BH4)1.6(AlH4)0.4 |
5.1 |
10.1 |
| Mg(BH4)1.6I0.4 |
|
2.8 |
The distance between Al–H (1.6 Å) is longer than between B–H (1.2 Å). The B–Al and B–B distances are 4.4 and 3.9 Å, respectively, in both LT and HT phases. Larger-sized ions offer a larger space for Mg atoms to move through the triangle face made by B and Al atoms. For the LT phase, a H–H exchange reaction between the different BH4 and AlH4 groups is partly observed when the two H atoms are shared by B and Al atoms.
The Mg atoms in Mg(BH4)1.6(AlH4)0.4 are coordinated not only by H atoms of BH4 and AlH4 of the tetrahedral cage surrounding that Mg atom but also those of the neighboring tetrahedral cages. Coordination including such H atoms is represented in Fig. 5 as a transparent cloud. The Mg atom in the center region is coordinated only by H atoms of the tetrahedral cage surrounding that Mg atom, as indicated by the solid surface in the figure. For pure Mg(BH4)2 and Mg(BH4)1.6 Cl0.4, almost no coordination from the H atoms of such neighboring cages is found as can been seen in Fig. 2 and 4, in which only a small transparent region is observed at the center. Thus, we can understand that stabilization by H atoms enables Mg atoms to move outside of the cage. The transition state as the atom passes is stabilized in the same manner as for Li+ conduction in LiBH4.7 There are large free spaces outside of the tetrahedral cages. When a Mg atom is forced to move into such spaces manually, it returns immediately to its original position due to the lower H-coordination of these spaces.
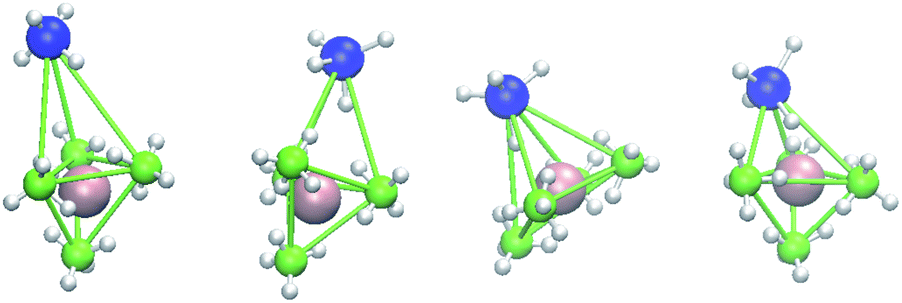
As illustrated in Fig. 6, large Mg movements are also possible from large cage deformations, in which two tetrahedral cages combine to form a hexahedral cage after deformation. A Mg atom located in one of the two tetrahedral cages moves to the center of the hexahedral cage. If this kind of change takes place continuously, Mg ions can diffuse within the crystal.
 |
| | Fig. 6 Change from tetrahedral to hexahedral cages in the HT phase of Mg(BH4)1.6 (AlH4)0.4. Figures from left to right are taken at the 0, 2067, 12510, 15423th step of the same atom group. At 12510th step, the lower tetrahedral cage is almost in plane. Green ball, B; large blue ball, Al; centered pink ball, Mg; small white ball, H. | |
3.4 Which is more important, size or H-coordination?
AlH4 has two features: large size and high H-coordination number. To separate these two effects, we have tested the substitution by iodine atom for BH4. I− has a large ionic radius of about 2.2 Å. The Mg atom and H-coordination number distributions are shown in Fig. 7. A RDF is given in Fig. 2(d). Even though the I− ion is large, the Mg atom is confined in the tetrahedral cage with no large displacement.
 |
| | Fig. 7 Mg- and H-atom distributions in the tetrahedral cage in Mg(BH4)1.6I0.4. Different anions, BH4 or I, are randomly placed at each apex of the tetrahedron. See note in Fig. 2. | |
While the substitution of AlH4 for BH4 has a good influence on the Mg movement, stability of the substituted compound is uncertain. We therefore performed a structure optimization of Mg(BH4)1.6 (AlH4)0.4, which gave a higher energy than (0.8)Mg(BH4)2 + (0.2)Mg(AlH4)2 by about 0.17 eV. AlH4 partial substitution of Mg(BH4)2 has not been reported at present, though Cl substituted Mg(BH4)2 was reported.35 Therefore, it is necessary to find a way to stabilize the substituted Mg(BH4)2,36 or to find another substitution like NH2. The last substituted one has already been produced37,38 and showed high Mg conductivity.3
4 Conclusion
In conclusion, Mg distribution in Mg(BH4)2 spreads beyond the tetrahedral cage surrounding the Mg atom by partly substituting a large hydride complex, AlH4, for BH4. We can expect that the transition state of the Mg ion transfer is stabilized by retaining high H-coordination from BH4 and AlH4 outside the tetrahedral cage. Controlling the H-coordination number might produce the higher Mg ion conductors. It is necessary to find a hydride resulting in a stable substitution to create a high Mg ion conductor.
Acknowledgements
Numerical calculations were performed on the K computer at the RIKEN Advanced Institute for Computational Science (Proposal number hp120146) and the PC clusters at the Research Institute for Information Technology, Kyushu University. This work was supported in part by JSPS KAKENHI (Grant no. 25220911), the Computational Materials Science Initiative (CMSI), Japan, and the Inter-university Cooperative Research Program of the Institute for Materials Research, Tohoku University. 3D images were generated using VMD.39
References
- S. Ikeda, M. Takahashi, J. Ishikawa and K. Ito, Solid State Ionics, 1987, 23, 125 CrossRef CAS.
- K. Kawamura, K. Morota, N. Nakamura, H. Maekawa, T. Hattori, N. Imanaka, Y. Okazaki and G. y. Adachi, Solid State Commun., 2001, 120, 295 CrossRef.
- S. Higashi, K. Miwa, M. Aoki and K. Takeuchi, Chem. Commun., 2013 10.1039/C3CC47097K.
- M. Matsuo, Y. Nakamori, S. Orimo, H. Maekawa and H. Takamura, Appl. Phys. Lett., 2007, 91, 224103 CrossRef.
- T. Ikeshoji, E. Tsuchida, K. Ikeda, M. Matsuo, H.-W. Li, Y. Kawazoe and S. Orimo, Appl. Phys. Lett., 2009, 95, 221901 CrossRef.
- T. Ikeshoji, E. Tsuchida, T. Morishita, K. Ikeda, M. Matsuo, Y. Kawazoe and S. Orimo, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 144301 CrossRef.
- T. Ikeshoji, Y. Ando, M. Otani, E. Tsuchida, S. Takagi, M. Matsuo and S. Orimo, Appl. Phys. Lett., 2013, 103, 133903 CrossRef.
- Y. Filinchuk, R. Cěrný and H. Hagemann, Chem. Mater., 2009, 21, 925–933 CrossRef CAS.
- V. Ozolins, E. H. Majzoub and C. Wolverton, Phys. Rev. Lett., 2008, 100, 135501 CrossRef CAS.
- Z. Łodziana and M. J. van Setten, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 024117 CrossRef.
- M. J. van Setten, G. A. de Wijs, M. Fichtner and G. Brocks, Chem. Mater., 2008, 20, 4952–4956 CrossRef CAS.
- A. Bil, B. Kolb, R. Atkinson, D. G. Pettifor, T. Thonhauser and A. N. Kolmogorov, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 224103 CrossRef.
- R. Caputo, A. Kupczak, W. Sikora and A. Tekin, Phys. Chem. Chem. Phys., 2013, 15, 1471 RSC.
- D. Blanchard, J. B. Maronsson, M. D. Riktor, J. Kheres, D. Sveinbjörnsson, E. G. Bardají, A. Leon, F. Juranyi, J. Wuttke, K. Lefmann, B. C. Hauback, M. Fichtner and T. Vegge, J. Phys. Chem. C, 2012, 116, 2013–2023 CAS.
- J. Voss, J. S. Hummelshoj, Z. L. odziana and T. Vegge, J. Phys.: Condens. Matter, 2009, 21, 012203 CrossRef CAS PubMed.
- J.-H. Her, P. W. Stephens, Y. Gao, G. L. Soloveichik, J. Rijssenbeek, M. Andrus and J.-C. Zhao, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 561–568 CAS.
- R. Cerny, Y. Filinchuk, H. Hagemann and K. Yvon, Angew. Chem., 2007, 119, 5867–5869 CrossRef.
- H. Maekawa, M. Matsuo, H. Takamura, M. Ando, Y. Noda, T. Karahashi and S. Orimo, J. Am. Chem. Soc., 2009, 131, 894 CrossRef CAS PubMed.
- H. Oguchi, M. Matsuo, T. Sato, H. Takamura, H. Maekawa, H. Kuwano and S. Orimo, J. Appl. Phys., 2010, 107, 096104 CrossRef.
- M. Matsuo, H. Oguchi, T. Sato, H. Takamura, E. Tsuchida, T. Ikeshoji and S. Orimo, J. Alloys Compd., 2013, 580, S98–S101 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703 CrossRef CAS.
- C. Hartwigsen, S. Goedecker and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 3641 CrossRef CAS.
- E. Tsuchida and M. Tsukada, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 7602 CrossRef CAS.
- E. Tsuchida and M. Tsukada, J. Phys. Soc. Jpn., 1998, 67, 3844 CrossRef CAS.
- F. Gygi, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 11190 CrossRef CAS.
- D. C. Liu and J. Nocedal, Math. Program., 1989, 45, 503 CrossRef.
- E. Tsuchida, J. Phys. Soc. Jpn., 2002, 71, 197 CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, R558 CrossRef.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater.
Phys., 1996, 54, 11169 CrossRef CAS.
- E. Tsuchida, J. Chem. Phys., 2011, 134, 044112 CrossRef PubMed.
- A. Fossdal, H. Brinks, M. Fichtner and B. Hauback, J. Alloys Compd., 2005, 387, 47–51 CrossRef CAS PubMed.
-
G. Giancovazzo, H. L. Monaco, G. Artioli, D. Vitergo, G. Ferraris, G. Gilli, G. Zanotti and M. Catti, Fundamentals of Crystallography, Oxford University Press, New York, 2nd edn, 2002 Search PubMed.
- A. Hino, J. E. Fonnel op, M. Corno, O. Zavorotynska, A. Damin, B. Richter, M. Baricco, T. R. Jensen, M. H. S. orby and B. C. Hauback, J. Phys. Chem. C, 2012, 116, 12482–12488 Search PubMed.
- S. Takagi, T. Ikeshoji, M. Matsuo, T. Sato, H. Saitoh, K. Aoki and S. Orimo, Appl. Phys. Lett., 2013, 103, 113903 CrossRef.
- T. Noritake, K. Miwa, M. Aoki, M. Matsumoto, S. Towata, H.-W. Li and S. Orimo, Int. J. Hydrogen Energy, 2013, 38, 6730 CrossRef CAS PubMed.
- T. Noritake, K. Miwa, M. Aoki, M. Matsumoto, S. Towata, H.-W. Li and S. Orimo, J. Alloys Compd., 2013, 580, S85 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Molec. Graphics, 1996, 14, 33 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.