 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of selective, ultra-fast multiple co-sensitization to control dye loading in dye-sensitized solar cells†
Peter J.
Holliman
*a,
Kareem J.
Al-Salihi
a,
Arthur
Connell
a,
Matthew L.
Davies
a,
Eurig W.
Jones
a and
David A.
Worsley
b
aSchool of Chemistry, Bangor University, Gwynedd LL57 2UW, UK. E-mail: p.j.holliman@bangor.ac.uk; Fax: +44 (0)1248 370528; Tel: +44 (0)1248 382375
bSPECIFIC, College of Engineering Swansea University, Baglan Bay Innovation and Knowledge Centre, Port Talbot SA12 7AZ, UK
First published on 4th November 2013
Abstract
Enhancing the spectral response of dye-sensitized solar cells (DSC) is essential to increasing device efficiency and a key approach to achieve this is co-sensitization (i.e. the use of multiple dyes to absorb light from different parts of the solar spectrum). However, precise control of dye loading within DSC mesoporous metal oxide photo-anodes is non-trivial especially for very rapid processing (minutes). This is further complicated by dyes having very different partition (Kd) and molar extinction (ε) coefficients which strongly influence dye uptake and spectral response, respectively. Here, we present a highly versatile, ultra-fast (ca. 5 min) desorption and re-dyeing method for dye-sensitized solar cells which can be used to precisely control dye loading in photo-electrode films. This method has been successfully applied to re-dye, partially desorb and re-dye and selectively desorb and re-dye photo-electrodes using examples of a Ru-bipy dye (N719) and also organic dyes (SQ1 and D149) giving η up to 8.1% for a device containing the organic dye D149 and re-dyed with the Ru dye N719. The paper also illustrates how this method can be used to rapidly screen large numbers of dyes (and/or dye combinations) and also illustrates how it can also be used to selectively study dye loading.
In 1991, O'Regan and Grätzel described a breakthrough increase in dye-sensitized solar cell (DSC) efficiency by sensitizing nanoparticulate TiO2 with Ru-bipyridyl dye.1 Since this, many new dyes have been reported2 with excellent spectral response from 450–600 nm where solar intensity is highest leading to η > 12%.3 However, there remains great scope to increase DSC efficiency and spectral response with two broad approaches being possible; either pan-chromatization using one dye with a wide spectral response or co-sensitization using multiple dyes to absorb at different wavelengths. Panchromatic dyes include Ru-terpyridyl “black dye” (η = 10%)4 and the squaraine (JK-216, η = 6.3%) dye5 although broader absorption can limit Voc because, in this approach, the LUMO must drop to ensure a long wavelength absorption onset and/or ε because it is difficult to design a dye with consistently high ε across the entire solar spectrum. By comparison, co-sensitization uses more than one dye so HOMO–LUMO energy levels can be off-set and dye loading can be used to help manage ε and spectral response. Previous co-sensitization examples include a multi-layer approach sensitizing TiO2 with one dye, then depositing an Al2O3 layer and then a second dye6 leading to η = 8.65% for JK2, Al2O3 and then SQ1.7 Other approaches have used supercritical fluids,8 specialist photo-electrodes9,10 or slow dyeing procedures11–15 to control dye uptake leading to the highest reported DSC efficiency (η > 12%) using a Zn-porphyrin and a triphenylamine dye with a Co complex as the redox couple.3
Current limitations on the co-sensitization approach include the dyeing times (i.e. hours) and controlling the dyeing process (e.g. dye loading and surface organisation). In this context, we recently reported ultra-fast co-sensitization (5 min) using N719 and SQ1 giving η = 7.9% (ref. 16) and the first example of ultra-fast tri-sensitization17 by pumping dye solution through pre-sealed devices to overcome the mass transfer issues of passive dyeing. Thus, ultra-fast sensitization is controlled by the rate of dye adsorption which is influenced by dye partition coefficients (solvated vs. adsorbed). However, these partition coefficients are fixed for each dye–solvent–metal oxide system which limits the control of dye loading in what is a competitive sorption process. Thus, increasing dye loading for a single dye involves simply dyeing for longer and rinsing away any excess. However, for multiple dyeing, this approach is only possible if all the dyes being used have identical partition coefficients which is extremely unlikely. Hence, dye uptake from a multiple dye solution is dominated by the highest partition coefficient dye.
It is well known that dye loading can be measured by dye desorption typically using NaOH to de-esterify chemisorbed dyes from the TiO2 surface. In general, desorption processes have been much less studied to date with Park et al. positioning dyes using gelled alkali18 and O'Regan et al. using dilute alkali to desorb dyes to study recombination processes.19
We report here new, ultra-fast co-sensitization procedures which overcome the limitations caused by varying ε and Kd between different dyes and which enable much greater control of dye loading even at processing times of minutes. This has been achieved through total, partial and/or selective dye desorption and re-dyeing of selected metal complex and organic dyes and replacement with others. This paper also reports links between Jsc, spectral response and device efficiency for multiply co-sensitized DSC devices including the first ever examples of ultra-selective, ultra-fast desorption of tri-sensitized DSC photo-electrodes. The data show that these approaches to desorption and re-dyeing are entirely tunable to different dyes and can produce bespoke DSC devices where the colour, spectral response and transparency etc. can be precisely controlled in a matter of minutes.
Results and discussion
Ultra-fast desorption by various alkali solutions
Different alkaline solutions have been tested for the ultra-fast desorption of N719 dye which had been adsorbed using ultra-fast pump dyeing (1 ml, 1 mM N719 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH3CN:tBuOH). A device was dyed, desorbed and then re-dyed after each desorption and IV data were measured at each stage (Table 1). The alkalis were chosen because they have similar pKb values with only a variance in the cation with the exception of tris(hydroxymethyl)ethylamine or THMA which is a weaker base (pKb = 8.0).
:1 CH3CN:tBuOH). A device was dyed, desorbed and then re-dyed after each desorption and IV data were measured at each stage (Table 1). The alkalis were chosen because they have similar pKb values with only a variance in the cation with the exception of tris(hydroxymethyl)ethylamine or THMA which is a weaker base (pKb = 8.0).
| Device | η/% | V oc/V | J sc/mA cm−2 | FF |
|---|---|---|---|---|
| a THMA = tris(hydroxymethyl)ethylamine. b pKb values are shown in parentheses. | ||||
| Dyed with N719 | 4.9 | 0.70 | 11.04 | 0.64 |
| Desorbed by KOH (−0.3)b | 0.3 | 0.58 | 0.74 | 0.55 |
| Re-dyed with N719 | 5.8 | 0.75 | 12.18 | 0.63 |
| Desorbed by NaOH (0.5) | 0.3 | 0.57 | 0.80 | 0.58 |
| Re-dyed with N719 | 5.3 | 0.72 | 12.21 | 0.61 |
| Desorbed by LiOH (0.2) | 0.2 | 0.53 | 0.81 | 0.48 |
| Re-dyed with N719 | 5.7 | 0.72 | 12.51 | 0.63 |
| Desorbed by Bu4NOH (0.3) | 0.3 | 0.56 | 0.82 | 0.60 |
| Re-dyed with N719 | 5.9 | 0.73 | 13.14 | 0.61 |
| Desorbed by THMAa (8.0) | 0.3 | 0.57 | 0.99 | 0.58 |
| Re-dyed with N719 | 6.0 | 0.73 | 13.26 | 0.62 |
The IV data in Table 1 show that, for the dyed devices, Jsc varies from 11.04 mA cm−2 for the initial N719 dyeing up to 13.26 mA cm−2 after the fifth cycle of dyeing. This increase might either be due to the changing cations or to the repeated pH cycling from high to low which could increase the number of available dye sorption sites presumably by increasing surface OH groups on the TiO2. Dyeing and desorbing an electrode using only Bu4NOH also shows an increase in η (from 4.6% to 5.0%) and increased N719 loading from 156 to 174 μg cm−2 suggesting changes in the dyeing sites outweigh any cation effects. By comparison, Table 1 also shows only a slight increase in Voc from 0.70 to 0.73 V and the FF remains essentially the same (0.64–0.62).
The desorbed devices all show similar and much lower Jsc (0.74–0.99 mA cm−2), Voc (0.58–0.57 V) and FF (0.55–0.58) in line with complete dye desorption (dye loading was ca. 162 μg N719 cm−2). These data show a cycle of highs and lows of device efficiency corresponding to dyed and desorbed devices which show an increase in device performance from the first (η = 4.9%) to the fifth cycle of dyeing (η = 6.0%) in line with Jsc. Corresponding images (ESI Fig. S1†) confirm the presence and absence of adsorbed dye during each dyeing, desorption and re-dyeing cycle. These data confirm that it is possible to ultra-fast dye, desorb and re-dye TiO2 photo-electrodes with N719 using a range of different alkaline solutions without apparently damaging any of the exposed device components. However, further studies show that whilst alkali metal hydroxides (e.g. NaOH or KOH) desorb Ru(bipy) complexes at room temperature that the removal of organic dyes such as SQ1 requires the dismantling of the device by re-heating the Surlyn gasket and removing the counter electrode followed by heating at ≥40 °C for 30 min which causes irreversible damage to device components.20 By comparison, Bu4NOH removes all dyes at room temperature without damaging device components and hence this base was chosen for more detailed studies.
Studies of ultra-fast dye desorption using Bu4NOH
Initially, the effects of Bu4NOH concentration (Table 2) and volume (Fig. 1) were trialled to study ultra-fast partial dye desorption and re-dyeing. Initially, a constant volume of 100 μl of Bu4NOH was used (Table 1). Given that a 25 μm Surlyn gasket was used and the photo-electrode area was ca. 1 cm2 (0.5 × 2.0 cm2) this suggests the device cavity was ca. 20 μl which means that the alkali would replenish this volume five times during the desorption cycle. The data show that this device gives η = 4.7–5.0% when fully dyed and that 20–40 mM Bu4NOH is sufficient to desorb all the N719. By comparison, 10 mM Bu4NOH results in partial dye desorption giving a ca. 4/5th drop in the Jsc value (2.2 mA cm−2versus 11.3 mA cm−2). Dropping this concentration to 4 mM results in only a small drop in Jsc (10 mA cm−2versus 11.3 mA cm−2) implying partial dye desorption is possible.| Device | η/% | V oc/V | J sc/mA cm−2 | FF |
|---|---|---|---|---|
| Dyed with N719 | 4.9 | 0.80 | 12.05 | 0.51 |
| Desorbed by 40 mM | 0.3 | 0.59 | 0.81 | 0.68 |
| Re-dyed with N719 | 5.0 | 0.77 | 11.87 | 0.55 |
| Desorbed by 20 mM | 0.4 | 0.6 | 0.85 | 0.69 |
| Re-dyed with N719 | 4.9 | 0.77 | 11.30 | 0.57 |
| Desorbed by 10 mM | 0.9 | 0.64 | 2.21 | 0.64 |
| Re-dyed with N719 | 4.7 | 0.76 | 11.26 | 0.55 |
| Desorbed by 4 mM | 4.2 | 0.76 | 10.02 | 0.56 |
| Re-dyed with N719 | 4.9 | 0.77 | 10.35 | 0.61 |
 | ||
| Fig. 1 η, Jsc and dye loading for a TiO2 device fast dyed with N719 (1 ml, 2.8 mM) and desorbed with varying volumes of 4 mM Bu4NOH. Efficiency shown as black squares and Jsc as open circles. Inset shows dye loading versus volume of Bu4NOH. | ||
The effects of Bu4NOH volume on N719 desorption have been studied between 1000 and 100 μl using a 4 mM Bu4NOH solution. Fig. 1 shows that higher volumes desorb more dye which reduces both Jsc and η in parallel as expected. However, the relationships between Bu4NOH volume, the loading of dye desorbed and the η/Jsc are not linear. Thus, 500–1000 μl of Bu4NOH desorbs between 121 and 132 μg cm−2 which drops to ca. half the initial dye desorption (71 μg cm−2) for 100 μl of alkali. By comparison, Jsc and η did increase in step-wise increments as the alkali volume drops. This suggests that the link between dye loading and Jsc when dye is desorbed from a pre-dyed device is more complicated than a simple linear relationship. Thus, it is known that the uppermost portion of a mesoporous film dyes more rapidly than the rest of the film (as this comes into contact with the dye solution first) and that ultra-fast dyeing overcomes the limitations of mass transport through such films to speed up complete dyeing of the film.16 In the same way, ultra-fast desorption is expected to overcome mass transport limitations of alkali solution passing through the film. However, changing the volume of the solution passing through the film must also changes the residence time (for a similar pumping rate) and this would be expected to have a greater impact on desorption rates than a simple linear proportionality. Overall, these data show that it is possible to partially remove adsorbed dye from a photo-electrode in a controlled manner by varying the volume and/or concentration of the dye solution. These data also suggest that some dye can be removed with relatively less impact on device performance. This is important because this should make dye sites available within the TiO2 for a different dye which, if it can harvest photons which are not absorbed by the first dye (due to poor spectral response at a particular wavelength) then this could potentially enhance overall device light harvesting.
Ultra-fast total desorption and re-dyeing using different dyes
Complete dye desorption and re-dyeing was then studied using a range of different dyes. In the first example, Device A1 was pump dyed with 1 ml of the Ru-bipy dye N719 giving η of 6.2% which is typical for a 1 cm2 TiO2 photo-electrode (Table 3). The N719 was then rapidly and completely desorbed by pumping 40 mM Bu4NOH(aq) through the device cavity. Complete dye removal was confirmed visually because the photo-electrode became white (Fig. 2) and an N719-coloured desorption solution was produced which was subsequently confirmed by UV-vis spectroscopy. In line with dye desorption, after re-filling with I3−/I− electrolyte, the device efficiency had dropped to η = 0.3%. After electrolyte removal and device cavity rinsing with 100 μl of ethanol, the device was re-dyed with N719 and the resulting Device A2 showed identical efficiency to the first dyeing (Device A1) with a small increase in Jsc which might suggest a slightly higher dye loading but also a 20 mV reduction in Voc. The slight increase in Jsc was further confirmed by the EQE data (Fig. 3) of the re-dyed device which shows a slightly higher EQEmax at ca. 560 nm (68%) compared to the initial dyeing (62%). This confirms that, when BuN4OH base is used for dye desorption, rapid and complete dye removal is possible at room temperature with no degradation of the metal oxide surface or any of the device components (conducting layers, platinized counter electrode) which are all also exposed to the desorption solution.| η/% | V oc/V | J sc/mA cm−2 | FF | ||
|---|---|---|---|---|---|
| A1 | Dye N719 | 6.2 | 0.72 | 13.45 | 0.64 |
| A2 | Re-dyed with N719 | 6.2 | 0.70 | 14.16 | 0.63 |
| B1 | Dye SQ1 | 3.1 | 0.59 | 8.21 | 0.64 |
| B2 | Re-dyed with SQ1 | 3.2 | 0.61 | 8.42 | 0.62 |
| C1 | Dye SQ1 | 3.1 | 0.59 | 8.31 | 0.62 |
| C2 | Re-dyed with N719 | 6.3 | 0.73 | 13.47 | 0.64 |
| D1 | Dye N719 | 6.0 | 0.71 | 13.22 | 0.64 |
| D2 | Re-dyed with SQ1 | 2.8 | 0.61 | 7.27 | 0.63 |
| E1 | Dye N719:SQ1 | 7.2 | 0.74 | 15.99 | 0.60 |
| E2 | Re-dyed – N719:SQ1 | 6.7 | 0.73 | 14.52 | 0.63 |
 | ||
| Fig. 2 DSC devices showing colour changes (a) before dyeing, (b) after N719 dyeing, (c) after N719 desorption – the yellow colour is due to I3−/I− electrolyte, (d) after N719 re-dyeing, (e) after partial desorption of N719 and (f) after re-dyeing with SQ1. | ||
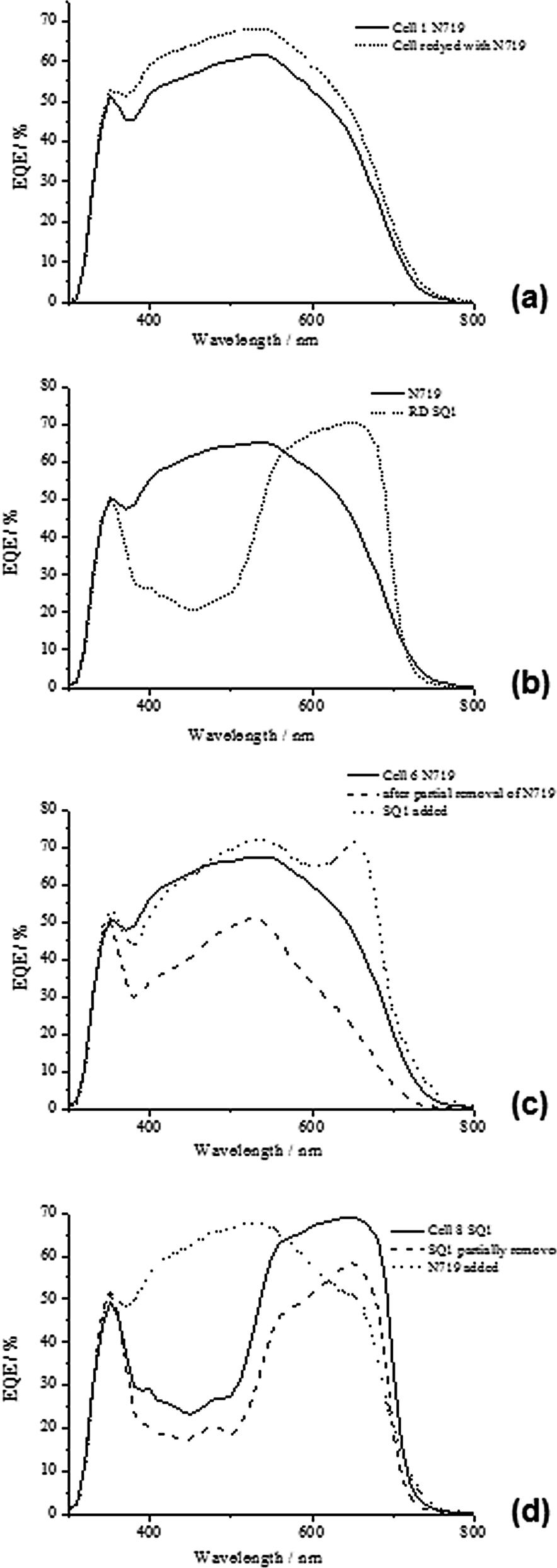 | ||
| Fig. 3 EQE data for dyed, desorbed and re-dyed DSC devices; (a) dyed and re-dyed N719, (b) dyed with N719, re-dyed with SQ1, (c) dyed with N719, partially removed and re-dyed with SQ1 and (d) dyed with SQ1, partial SQ1 removal and re-dyed with N719. | ||
Device B1 was dyed with 300 μl of ethanolic SQ1 solution; the lower volume of solution reflecting the much higher ε of SQ1 compared to N719 (ca. 158500 (ref. 21) vs. ca. 14000 M−1 cm−1 (ref. 22)). The data (Table 3) show a similar pattern to N719 with the initial SQ1 dyeing (Device B1) giving η = 3.1% which dropped to 0.3% after dye desorption with 40 mM Bu4NOH. Again the desorbed photo-electrode became white and the desorbed dye solution became SQ1-coloured (blue) which was confirmed by UV-visible spectroscopy. Re-dyeing with SQ1 again gave almost identical device performance (Device B2) to the initial SQ1 dyeing although very slight increases in Voc (20 mV) and Jsc (0.2 mA cm−2) were observed which again suggests that no degradation of the device components had taken place. Again the EQEmax of the re-dyed device at ca. 650 nm showed a slight increase compared to the initial dyeing although this is balanced by a small decrease in EQE between 400 and 500 nm where the dye is less efficient which may explain why the increase in Jsc increase is small (see ESI†).
Having established that one dye can be removed and replaced without any loss of performance, a study was carried out to see if the same photo-electrode could be desorbed and dyed with different dyes. Thus, Device C1 was first dyed with 300 μl of SQ1 solution showing η of 3.1% which is in line with the data for Device B1. After removal of this dye, the same device was dyed with 1 ml of N719 and the efficiency increased to 6.3% (Device C2); reflecting an increase in Jsc and Voc for N719 versus SQ1. The data for Device C2 are also in line with the data for Device A1 with almost identical Jsc and Voc again support the idea that no change had occurred to the device components during desorption and re-dyeing. To test whether the order of dyeing affects the process, Device D1 was first pump dyed with N719 giving η of 6.0% which is comparable to the other N719 devices. However, Device D2 shows a drop in η to 2.8% after N719 desorption and re-dyeing with SQ1 which was due to lower Jsc and Voc typical for SQ1 vs. N719. The EQE data for Device D1 (Fig. 3b) confirms the differing EQE responses for the different dyes with an initial broad spectral response (EQEmax = 560 nm) typical of N719 followed by the typical response for SQ1 (EQEmax = 640 nm). These data confirm that the N719 dye is desorbed and that only SQ1 dye is responsible for light harvesting in the re-dyed device.
Having established that it is possible to change dyes within the same device, a study was carried out to test if a co-sensitized device can be desorbed and re-dyed. To do this, Device E1 was dyed with 1 ml of a mixed dye solution containing 2.8 mM N719 and 0.17 mM SQ1 giving η = 7.2% (Table 3) which is greater than the efficiency for N719 alone (Device A1) mainly due to an increase in Jsc as might be expected for an increased spectral response from co-sensitization. When the dyes are desorbed using 40 mM Bu4NOH, all the dyes are fully removed as evidenced by a change in colour of the photo-electrode to white whilst UV-visible spectroscopy confirms an initial dye loading of 202.0 and 1.0 μg cm−2 for N719 and SQ1, respectively.
After addition of electrolyte, the device efficiency drops (η = 0.3%) in line with complete dye removal. After washing and re-dyeing with 1 ml of the same N719:SQ1 dye solution, the device efficiency returns to η = 6.7% (Device E2); the drop from Device E1 being due to a drop in Jsc is reflected in the slightly lower EQE (see ESI†) although the signal for SQ1 is clearer for the re-dyed device presumably due to the weaker response of N719. Further dye desorption of this device actually shows a higher N719 loading (240 μg cm−2) but a similar SQ1 loading. This might possibly suggest some slight N719 dye aggregation causing reduced light harvesting. Whilst the data in Table 3 confirm that dyeing, desorption and re-dyeing can be successfully achieved for single and mixed dye solutions and the consistent fill factors suggest that device components are not damaged by careful choice of desorption and rinsing solutions even over many cycles (data not shown), Device E suggests that it can be more difficult to precisely control dye loadings from mixed dye solutions.
Partial dye desorption and re-dyeing
To further study the ability to accurately control dye loading in multiply dyed devices, partial dye desorption and re-dyeing with a different dye was first studied. To do this, Device F1 was first dyed with 1 ml of N719 solution giving a typical efficiency for this dye (η = 6.5%, Table 4). Then N719 desorption was studied using a small volume (100 μl) of a low concentration (1 mM) of Bu4NOH(aq). This resulted in partial N719 removal such that UV-vis spectroscopy showed that 80 μg cm−2 of N719 was desorbed and Jsc dropped from 13.19 to 12.29 mA cm−2 (Device F2). The EQE response after partial removal of N719 also showed a reduction in the EQEmax at ca. 550 nm from ca. 65% down to just under 50%. The Voc for Device F2 also dropped 50 mV to 0.69 V which may reflect increased back reaction taking place at vacated dye sites on the TiO2 surface. It is interesting to note that again the drop in Jsc and η are less than might be expected for a linear relationship between dye loading and η. This is an important result for several reasons. Firstly, it implies that there are some dye sites within the electrode which do not operate as efficiently as others. In turn, this implies that these vacated sites could be sensitized with a different dye which might absorb at a different part of the solar spectrum and so increase the overall photo-current. In the example here (Device F3, Table 4) this is exactly what happens when the device is re-dyed with SQ1 with a small increase in Jsc to 13.87 mA cm−2 although the Voc actually drops another 20 mV to 0.67 V which reflects the addition of SQ1 which has a lower Voc than N719 (see e.g. Device B1 in Table 3). This limits η to 5.6% for Device F3. EQE confirms the additional light harvesting of SQ1 and confirms that both dyes are contributing to the spectral response of this device (Fig. 3c). Interestingly, complete dye desorption from Device F3 gave dye loadings of 159 and 13 μg cm−2 for N719 and SQ1, respectively. Thus, Device F3 has a higher SQ1 loading than Device E which, by comparison, was co-sensitized from a mixed N719:SQ1 dye solution under conditions of directly competitive adsorption between these two dyes. The EQE signal for SQ1 is much clearer in Device F3 than in Device E in line with the higher SQ1 loading.| Device | η/% | V oc/V | J sc/mA cm−2 | FF | |
|---|---|---|---|---|---|
| F1 | N719 | 6.5 | 0.74 | 13.19 | 0.67 |
| F2 | Partial desorption | 5.2 | 0.69 | 12.29 | 0.62 |
| F3 | SQ1 added | 5.6 | 0.67 | 13.87 | 0.60 |
| G1 | Dyed with SQ1 | 2.8 | 0.60 | 8.37 | 0.65 |
| G2 | Partial SQ1 removal | 2.2 | 0.56 | 6.29 | 0.63 |
| G3 | N719 added | 6.5 | 0.60 | 15.02 | 0.72 |
Looking at the dyeing, partial desorption and re-dyeing cycle for Device F, although it is difficult to conclusively prove the following effect, it is possible that the small volume of alkali used to partially desorb the N719 might cause preferential desorption of dye in the upper part of the photo-electrode. This might explain the non-linear relationship between dye loading and Jsc because presumably the most effective N719 should located closest to the direction of illumination. In these examples, which have used normal illumination, the dye closest to the working electrode should be the most effective. Hence, this might suggest that ultra-fast selective positioning of dyes within the photo-electrode is possible which is an important issue for scaling up the manufacture of these devices as previous reports have of dye positioning have required processing times of hours.8
Devices G1–G3 have been dyed in the opposite order to study how this affects device performance. Hence, Device G1 was first dyed with 500 μl of 0.70 mM SQ1 (with 5 mM CDCA) giving η = 2.8%. SQ1 was then partially removed using 10 μl of 1 mM BuN4OH giving η = 2.2% mainly due to a drop in Jsc in line with partial dye removal (Device G2). After rinsing the device cavity, the photo-electrode was re-dyed with 1 ml of N719 solution resulting in a large increase in Jsc from 6.29 to 15.02 mA cm−2 and an efficiency of 6.5%. This Jsc value is comparable to Device E2 which was also co-sensitized with a mixture of SQ1 and N719. The changing dye loadings are reflected in the EQE data (Fig. 3d) which show that the initial dyeing gives a typical SQ1 spectral response with EQEmax 68% at ca. 650 nm but that this response drops to just under 60% after partial dye desorption. However, re-dyeing with N719 shows a big change with the broad spectral response of N719 being apparent with EQEmax at 550 nm with a drop in the SQ1 response to ca. 50% which is a different effect from when the dyeing takes place in the opposite order in Device F (Fig. 3c) where the SQ1 signal is at 70% but appears narrower which may suggest less SQ1 aggregation. This would be logical because adding SQ1 into sites vacated by N719 would have to result in isolated SQ1 molecules whereas the opposite would occur if N719 is added afterwards.
The changing dye loadings were also observed in the changing colour of the photo-electrode after each dyeing step (see ESI†). In line with the lower dye loading very little alkali solution is required to remove some of the SQ1. It is interesting though how much re-dyeing with N719 increases the photo-current when initially it might be expected that there would be few dyeing vacant sites available. One possibility is that there might be different dyeing sites for these two dyes; one set of sites might not be available to SQ1 but remain available to N179. It is also possible that the small volume and low concentration of BuN4OH used could partially desorb the SQ1 in Device G2 preferentially from the “top” of the photo-electrode (closest to the holes in the counter electrode through which the desorption solution is pumped). This could leave relatively more vacant dye sites in this region of the photo-electrode. On this basis, re-dyeing with N719 might be expected to preferentially dye the “top” of the photo-electrode with N719. However, comparing the Devices F3 and G3, the data suggest that, for these two sensitizers, the order in which the dyes are adsorbed onto the TiO2 surface is more important than the position of the dyes within the photo-electrode. This may reflect more favourable surface organisation when SQ1 is allowed to adsorb before N719. However, the ability to selectively position dyes within a photo-electrode remains an important concept and is likely to be advantageous in a number of scenarios. For instance, where two or more dyes have vastly different partition coefficients (Kd), the dye with the larger Kd will always dominate the surface making precise control of dye loadings difficult. In addition, for multiply sensitized devices where the dye combinations used either suffer inter-dye recombination problems separating the dyes to different parts of the photo-electrode should reduce this problem.
Selective dye desorption
Having established that it is possible to partially desorb and re-dye DSC devices, a study of selective dye desorption was carried out to see if it is possible to multiply co-sensitize a TiO2 photo-electrode and then selectively remove different dyes. To do this, Device H1 was first dyed with 2 ml of a mixed dye solution containing 0.70 mM N719, 0.375 mM D149 and 10 mM CDCA giving η = 7.2% and a Jsc of 16.72 mA cm−2 which is significantly more than the photo-current achieved for N719 alone (e.g. Device A1). This result is interesting because both N719 and D149 absorb at similar wavelengths. However, the EQE data show that improved light harvesting can still be achieved because there is mismatch between the EQE profiles of the two dyes such that the correct dye loading of a mixture of both dyes should result in a broader EQE response than is possible for either dye on it's own (see ESI†). Such an effect has previously been shown for triarylamine dyes combined with N719 (ref. 16) and also for an organic dye with a porphyrin.3 After this, the N719 dye was selectively removed using 100 μl of 0.1 M LiOH(aq) resulting in a drop in η to 4.6% mainly due to the Jsc dropping to 11.40 mA cm−2 (Device H2). Selective N719 removal was confirmed by UV-vis spectroscopy (ESI Fig. S12†) which showed only N719 in the LiOH solution. Interestingly, when the device was re-dyed with 1 ml of standard N719 solution, the efficiency increased to 8.1% (Device H3) which was higher than that for the original device in this series (Device H1) mainly due to Jsc increasing to 17.8 mA cm−2 (Table 5). This again suggests that the method of dyeing can strongly affect device performance presumably because this influences the organisation of dyes on the TiO2 surface. This can be observed in the increased currents observed for the single dyes (Devices A1 and B1) but is even more apparent when more than one dye is present in the same photo-electrode (e.g. Device H3).| Device | η/% | V oc/V | J sc/mA cm−2 | FF | |
|---|---|---|---|---|---|
| H1 | D149:N719 | 7.2 | 0.69 | 16.72 | 0.69 |
| H2 | Partial N719 removal | 4.6 | 0.66 | 11.40 | 0.61 |
| H3 | Re-dyed with N719 | 8.1 | 0.71 | 17.81 | 0.64 |
| I1 | Dyed with N719 | 5.9 | 0.78 | 10.77 | 0.63 |
| I2 | SQ1 added | 5.1 | 0.74 | 9.25 | 0.67 |
| I3 | N719 desorbed | 0.7 | 0.61 | 1.42 | 0.68 |
| I4 | Re-dyed with SQ1 | 2.3 | 0.66 | 4.47 | 0.71 |
| I5 | N719 added | 5.2 | 0.73 | 9.75 | 0.66 |
| J1 | Dyed with N719 | 5.1 | 0.81 | 11.30 | 0.56 |
| J2 | Partial N719 removal | 3.6 | 0.63 | 10.16 | 0.57 |
| J3 | D149 added | 3.8 | 0.65 | 11.82 | 0.50 |
| J4 | Selective N719 removal | 2.9 | 0.60 | 8.27 | 0.58 |
| J5 | Re-dyed with N719 | 5.9 | 0.80 | 11.70 | 0.51 |
| K1 | Dyed with SQ1 | 2.3 | 0.60 | 6.39 | 0.61 |
| K2 | N719 added | 4.8 | 0.68 | 13.47 | 0.53 |
| K3 | D149 added | 5.5 | 0.69 | 13.36 | 0.59 |
| K4 | N719 desorbed | 0.8 | 0.78 | 1.44 | 0.67 |
| K5 | SQ1 desorbed | 1.3 | 0.61 | 3.72 | 0.60 |
| K6 | D149 desorbed | 0.3 | 0.57 | 0.84 | 0.62 |
The processes of dyeing, desorption and re-dyeing in dye sensitized solar cells rely on the fact that dyes are chemisorbed onto the metal oxide (TiO2) surface through ester linkages between dye carboxylate linker groups and surface hydroxyl groups of the metal oxide. The desorption step must therefore involve hydrolysis of the dye-surface ester bonds to release the dye molecules but without affecting either the dye or any of the other device components. Our evidence is that this can best be controlled through a combination of controlled alkalinity (OH−) and a suitable counter-ion (Bu4N+ or Li+). Hence these data show that it is possible to achieve completely selective dye desorption for N719 by using LiOH(aq). A further advantage of using Bu4NOH or LiOH as the base for desorption is that, after dye desorption, the Bu4N+ salt of the dye is formed which can be re-used to dye other photo-electrodes.
The step-wise co-sensitization and selective desorption of N719 and SQ1 have also been studied by first dyeing a photo-electrode with N719 (1 ml, 2.5 mM) giving Device I1 with η = 5.8% (Table 5). After electrolyte removal and rinsing, SQ1 (300 μl, 0.28 mM) was added was added to the N719-dyed electrode giving Device I2 with lower efficiency (5.1%) mainly due to Jsc decreasing from 10.77 mA cm−2 to 9.25 mA cm−2. Whilst the reason for the drop in Jsc is not entirely clear, it could be that no additional sorption sites are available as was the case for the partially desorbed Device F3 so the additional SQ1 may have aggregated with the N719 possibly causing some quenching of its excited state. N719 was then selectively desorbed by LiOH (50 μl, 100 mM) giving Device I3 where η had dropped to 0.65% which was due to only a small SQ1 loading remaining. The UV-vis spectrum for desorbed N719 by LiOH (see ESI†) gives two peaks at 504 nm and 364 nm corresponding to 63.7 μg N719 cm−2 and no evidence for SQ1 desorption. SQ1 was then desorbed using (Bu4NOH 100 μl, 4 mM). The resulting UV-vis spectrum shows only SQ1 corresponding to a loading of 11.8 μg SQ1 cm−2. After re-dyeing with SQ1 (300 μl, 0.28 mM), Device I4 gave η = 2.1% which increased to 5.2% when 1 ml of N719 (2.8 mM) was added (Device I5).
A similar experiment was carried out for N719 and D149 but using partial and selective desorption (Table 5). Thus, Device J1 was initially dyed with 1 ml of 1 mM N719 giving η of 5.1% followed by partial N719 desorption using Bu4NOH (50 μl, 4 mM) giving Device J2 with η = 3.6% and Jsc dropping to 10.16 mA cm−2 in line with the reduced N719 loading (UV-vis spectroscopy confirmed removal of 116 μg N719 cm−2). D149 (250 μl, 0.5 mM) was then added (Device J3) slightly increasing η to 3.8% and Jsc to 11.82 mA cm−2. However, Voc dropped to 0.65 V because D149 gives lower Voc than N719. Following this, 106 μg N719 cm−2 was selectively and completely desorbed by a larger volume of LiOH (200 μl, 100 mM) giving Device J4 which, as expected, lowered η to 2.9%, Jsc to 8.28 mA cm−2 and Voc to 0.60 V. The remaining D149 loading (22 μg D149 cm−2) was then desorbed by Bu4NOH (300 μl, 40 mM) followed by 0.5 ml acetone. After washing and rinsing, the device re-dyeing with N719 (1 ml, 1 mM), Device J5, and the η increased to 5.9% which may due the activation of TiO2 surface leading to form a monolayer of adsorbed dye molecules.
Sequential dyeing and desorption was then studied using SQ1, N719 and D149. First Device K1 was dyed with SQ1 (300 μl, 0.28 mM) giving η 2.3% which increased to 4.8% after 500 μl of 1 mM N719 was added giving Device K2 and 5.5% after 200 μl of 0.5 mM D149 was added giving Device K3. The Jsc increased in line with the addition of each dye in turn (Table 5). N719 was then selectively removed using LiOH (100 μl, 100 mM) giving Device K4 with η dropping to 0.8% and Jsc to 1.44 mA cm−2 suggesting that N719 was the dominant light harvesting dye in this device. UV-vis spectroscopy (ESI Fig. S12†) confirmed selective N719 removal by LiOH and a loading of 132.5 μg N719 cm−2 (Fig. 5). Next, SQ1 was selectively desorbed by Bu4NOH (100 μl, 1 mM) giving Device K5. This actually increased η to 1.3% with slightly increased Jsc (3.7 mA cm−2). The reason for this increase is not clear but presumably reflects the changing nature of the TiO2 surface with only D149 molecules adsorbed along with vacant sorption sites. UV-vis spectroscopy again confirmed selective SQ1 removal (Fig. 5) and a loading of 1.2 μg SQ1 cm−2. Finally, D149 was desorbed by Bu4NOH (100 μl, 40 mM) followed by 200 μl acetone and 100 μl ethanol. UV-vis spectroscopy confirmed that only D149 was desorbed giving a loading of 3.9 μg D149 cm−2. EQE of the tri-sensitized device showed responses for SQ1 at 660 nm and D149/N719 at ca. 550 nm (Fig. 4). The EQE for Device K4 containing SQ1 and D149 is significantly reduced after selective removed N719. The increase after selective SQ1 removal is in line with the I–V data discussed above (Device K5). Fig. 5 shows photographs of a tri-sensitized device showing a dark brown coloration followed the by the same device after selective desorption of each dye also showing the desorbed dye solutions showing the red-brown N719, the blue SQ1 and the pink D149.
 | ||
| Fig. 4 EQE data for (a) DSC device dyed with SQ1, then N719, then D149 and (b) DSC device dyed with N719 + SQ1 + D149 mixture and then step-wise selective desorption of N719, then SQ1, then D149. | ||
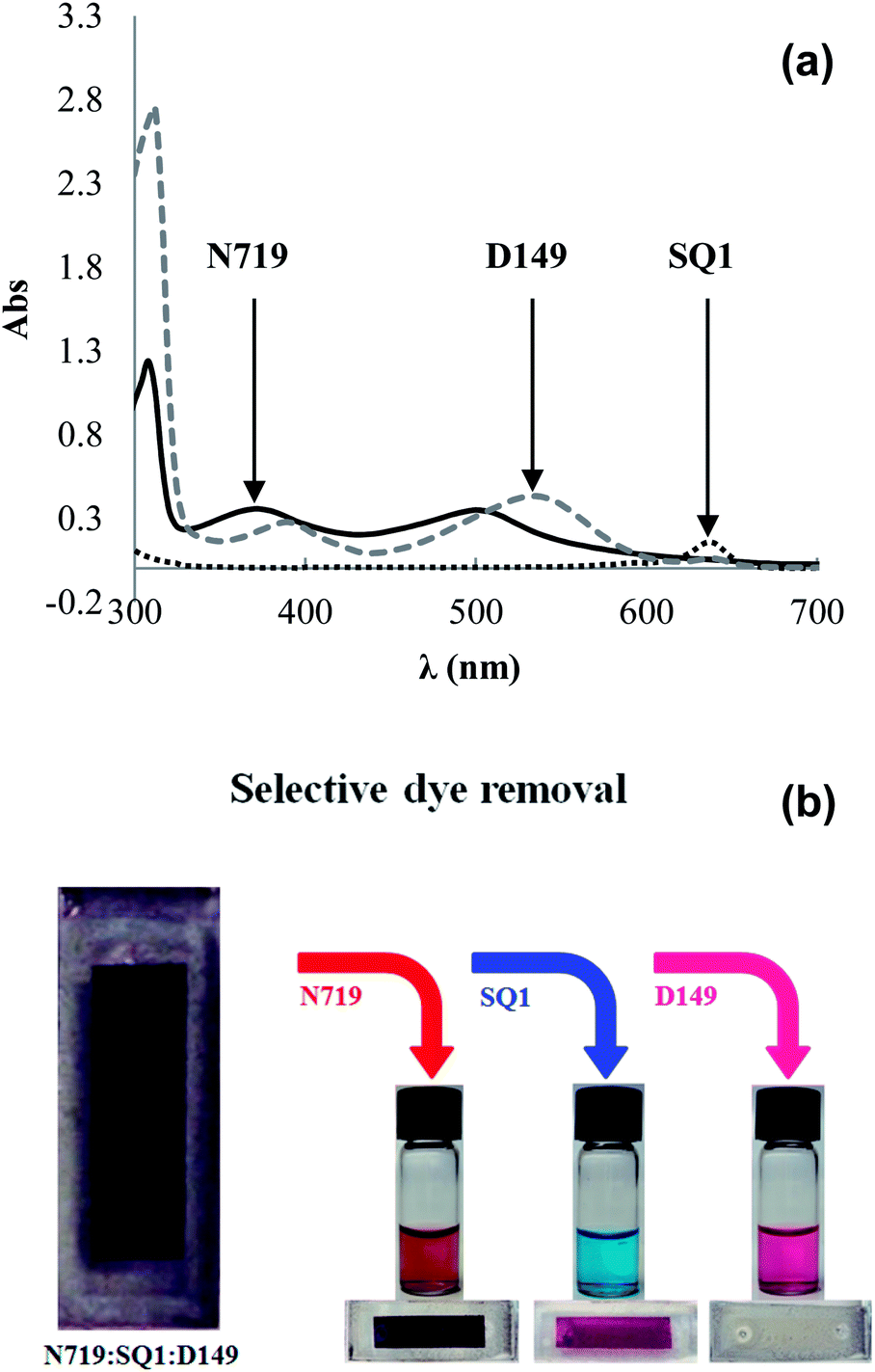 | ||
| Fig. 5 (a) UV-visible spectra of desorbed dyes and (b) photographs of co-sensitized DSC and the same device after step-wise, selective desorption of N719, SQ1 and D149. | ||
Conclusions
To make dye-sensitized solar cells more efficient, it is essential to significantly extend spectral response as far as possible from 400 to 700 nm. In practice, this means extending out into the longer wavelength region whilst maintaining the spectral response from 450 to 700 nm which is where the most efficient devices reported to date operate. To achieve this whilst maintaining a consistently high spectral response at all wavelengths is most likely to require the use of multiple dyes along with very close control of dye loadings and most probably dye positioning as well. Our data show that dye desorption and re-dyeing offers great control of all these parameters whilst also suggesting that it is potential to reduce the amount of dye used within a photo-electrode with relatively less effect on device performance with obvious benefits for reducing cost. The method can also be easily adapted for very rapid, high throughput testing of multiple dyes using a single device as we have repeated multiple dyeing–desorption–dyeing cycles through the same device cavities without any loss of device performance. In the context of the long-term stability of DSC devices, these data may also provide a warning about the use of electrolyte components such as tertiary-butyl pyridine which may have the potential to desorb dye in the presence of water if this is able to enter the device over extended weathering periods.Acknowledgements
We gratefully acknowledge Iraqi Govt. support for KJA, ERDF-WG LCRI funding for SPARC (AC, EWJ and MLD), EPSRC SPECIFIC funding (MLD) and NSG for supply of TEC™ glass to the SPARC consortium.Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629 CrossRef CAS PubMed.
- M. K. Nazeeruddin, P. Péchy and M. Grätzel, Chem. Commun., 1997, 1705 RSC.
- S. Pack, H. Choi, C. Kim, N. Cho, S. So, K. Song, M. K. Nazeeruddin and J. Ko, Chem. Commun., 2011, 47, 2874 RSC.
- J. N. Clifford, E. Palomares, M. K. Nazeeruddin, R. Thampi, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2004, 126, 5670 CrossRef CAS PubMed.
- H. Choi, S. Kim, S. O. Kang, J. Ko, M.-S. Kang, J. N. Clifford, A. Forneli, E. Palomares, M. K. Nazeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2008, 47, 8259 CrossRef CAS PubMed.
- F. Inakazu, Y. Noma, Y. Ogomi and S. Hayase, Appl. Phys. Lett., 2008, 93, 093304 CrossRef.
- R. Y. Ogura, S. Nakane, M. Morooka, M. Orihashi, Y. Suzuki and K. Noda, Appl. Phys. Lett., 2009, 94, 073308 CrossRef.
- K. Lee, S. W. Park, M. J. Ko, K. Kim and N. Park, Nat. Mater., 2009, 8, 665 CrossRef CAS PubMed.
- J.-J. Cid, J.-H. Yum, S.-R. Jang, M. K. Nazeeruddin, E. Martinez-Ferrero, E. Palomares, J. Ko, M. Grätzel and T. Torres, Angew. Chem., Int. Ed., 2007, 46, 8358 CrossRef CAS PubMed.
- D. Kuang, P. Walter, F. Nűesch, S. Kim, J. Ko, P. Comte, S. K. Zakeeruddin, M. K. Nazeeruddin and M. Grätzel, Langmuir, 2007, 23, 10906 CrossRef CAS PubMed.
- J. N. Clifford, A. Forneli, H. Chen, T. Torres, S. Tan and E. Palomares, J. Mater. Chem., 2011, 21, 1693 RSC.
- C.-M. Lan, H.-P. Wu, T.-Y. Pan, C.-W. Chang, W.-S. Chao, C.-T. Chen, C.-L. Wang, C.-Y. Lin and E. W.-G. Diau, Energy Environ. Sci., 2012, 5, 6460 RSC.
- L. H. Nguyen, H. K. Mulmudi, D. Sabba, S. A. Kulkarni, S. K. Batabyal, K. Nonomura, M. Grätzel and S. G. Mhaisalkar, Phys. Chem. Chem. Phys., 2012, 14, 16182 RSC.
- P. J. Holliman, M. L. Davies, A. Connell, B. V. Velasco and T. M. Watson, Chem. Commun., 2010, 46, 7256 RSC.
- P. J. Holliman, M. Mohsen, A. Connell, M. L. Davies, K. Al-Salihi, M. B. Pitak, G. J. Tizzard, S. J. Coles, R. W. Harrington, W. Clegg, C. Serpa, O. H. Fontes, C. Charbonneau and M. J. Carnie, J. Mater. Chem., 2012, 22, 13318 RSC.
- S. W. Park, K. Lee, D.-W. Lee, M. J. Ko, N.-G. Park and K. Kim, Nanotechnology, 2011, 22, 045201 CrossRef PubMed.
- B. O'Regan, L. Xiaoe and T. Ghaddar, Energy Environ. Sci., 2011, 5, 7203 Search PubMed.
- P. J. Holliman, N. Ahmed and M. Davies, unpublished work.
- J. H. Yum, P. Walker, S. Huber, D. Rentsch, T. Geigar, F. Nuesch, F. de Angelis and M. Grätzel, J. Am. Chem. Soc., 2007, 129, 10320 CrossRef CAS PubMed.
- P. Wang, C. Klein, R. Humphrey-Baker, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 808 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra42131g |
| This journal is © The Royal Society of Chemistry 2014 |