
Development of sustainable fluorous chemistry: the synthesis and characterization of fluorous ethers with nonafluoro-tert-butoxy groups†
See-Wing
Lo
a,
Edwin
Law
a,
Matthew Y.
Lui
a,
Xi-Guang
Wei
a,
Kai-Chung
Lau
*a,
Chui Y.
Ma
ab,
Margaret B.
Murphy
*ab and
István T.
Horváth
*a
aDepartment of Biology and Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong. E-mail: istvan.t.horvath@cityu.edu.hk; Fax: +852 3442-0522; Tel: +852 3442-7402
bState Key Laboratory in Marine Pollution, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong
First published on 13th October 2014
Abstract
Nonafluoro-t-butyl propyl (1), allyl (2), and propargyl (3) ethers as well as 1,2-bis(nonafluoro-t-butoxy)ethane (4), 1,3-bis(nonafluoro-t-butoxy)-propane (5), and 1,4-bis(nonafluoro-t-butoxy)-butane (6) were prepared by the reaction of sodium nonafluoro-t-butoxide (7) with the corresponding alkyl halides in good yields. Their fluorous partition coefficients and toxicity were also investigated. Computational studies have shown that 4, 5 and 6 exist in a monomeric form in MeOH–CF3C6F11 and they likely aggregate to form oligomers in PhCH3–CF3C6F11.
Introduction
Heterogenization of homogeneous catalysts has become a very attractive area of research in order to combine higher yields of homogeneous catalysts with facile separation of heterogeneous systems.1 One of the most popular heterogenized homogeneous catalytic systems involves two liquid phases with no, limited, or temperature-regulated miscibility of the two phases.1,2 While one of the phases is the catalyst phase containing the dissolved catalyst, the other is the product phase with no or limited solubility in the catalyst phase. The selection of the catalyst phase should be governed by the solvent properties of the substrate(s) and product(s).3 For example, in the case of apolar substrates and polar products, the best separation can be achieved by the application of fluorous4 or super critical carbon dioxide5 based biphasic systems.The fluorous biphasic concept was based on the limited miscibility of fluorous solvents with polar and/or protic organic media.3 Since higher fluorous partition coefficients (PF)3,6 or fluorophilicity (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) PF)7 of an organic compound can be achieved by increasing the fluorine content above 65%,6,7 the attachment of one or more C6–12-perfluoroalkyl chains to the hydrocarbon domains has been the typical design approach.2,4 Since fluorous compounds could be released to the environment, the oxidative fragmentation of their hydrocarbon domain could lead to C6–12-perfluoroalkylcarboxylates (PFCAs), which are persistent, have long half-lives in human and animal bodies, and may be toxic.8 The attachment of shorter C1–4-perfluoroalkyl-chains has been proposed9 to limit bioaccumulation and toxicity.10 The nona-fluoro-t-butoxy group, [(CF3)3C–O– or F9-t-butyl–O–)], is one of the shorter perfluoroalkyl chains that could provide high fluorous partitions6 and has been used to prepare fluorous aromatics, amines, azides, imidazoles, chiral carboxylic acids,11 and a fluorous Mitsunobu reagent.12
PF)7 of an organic compound can be achieved by increasing the fluorine content above 65%,6,7 the attachment of one or more C6–12-perfluoroalkyl chains to the hydrocarbon domains has been the typical design approach.2,4 Since fluorous compounds could be released to the environment, the oxidative fragmentation of their hydrocarbon domain could lead to C6–12-perfluoroalkylcarboxylates (PFCAs), which are persistent, have long half-lives in human and animal bodies, and may be toxic.8 The attachment of shorter C1–4-perfluoroalkyl-chains has been proposed9 to limit bioaccumulation and toxicity.10 The nona-fluoro-t-butoxy group, [(CF3)3C–O– or F9-t-butyl–O–)], is one of the shorter perfluoroalkyl chains that could provide high fluorous partitions6 and has been used to prepare fluorous aromatics, amines, azides, imidazoles, chiral carboxylic acids,11 and a fluorous Mitsunobu reagent.12
Results and discussion
We report the synthesis of F9-t-butyl propyl (1), F9-t-butyl allyl (2), and F9-t-butyl propargyl (3) ethers13 as well as 1,2-bis(F9-t-butoxy)ethane (4), 1,3-bis(F9-t-butoxy)-propane (5), and 1,4-bis(F9-t-butoxy)-butane (6) by the reaction of the corresponding alkyl halides with sodium F9-t-butoxide (7). The latter can be readily prepared from perfluoro-t-butanol (8). The fluorous ethers 1, 2, and 3 were isolated after 7 was treated with propyl bromide, allyl bromide, and propargyl chloride in 64.0, 36.0, and 56.0% yields, respectively (Fig. 1). It should be noted that a synthesis of 2 in 56% was recently reported.14 | ||
| Fig. 1 Synthesis of fluorous compounds 1–6. | ||
The fluorous diethers 4–6 were prepared by the reaction of the corresponding dihaloalkanes with 7 in DMF. Dibromoalkanes were preferred over dichloroalkanes, as the former were much more reactive. It was particularly difficult to displace both chloride atoms when the two nucleophilic centres were in close proximity. For example, 1,3-dichlorobutane could be converted to 6 but 1,2-dichloroethane did not react at all to form 4. At the end of the reactions, water was added to the reaction mixtures at 0 °C and the heavier lower fluorous phase was separated and subject to further purification. The isolated yields of 4–6 were in the range of 40–55%. While 3 is a white crystalline solid at room temperature, all the others were colourless liquids. The structures of compounds 1–6 were confirmed by MS, IR and NMR spectroscopy.
The fluorous partition coefficients of compounds 1–6 were measured in several fluorous/organic biphasic systems. Their concentrations were established in the lower fluorous and the upper organic phases of the 1:1 mixture of perfluoro-(methylcyclohexane) and several organic solvents including toluene, DMC and C1–4 alcohols by GC-MS analysis (Table 1).
| Cpdsa | Fluorine wt% | Fluorous partition coefficients (PF) | |||||
|---|---|---|---|---|---|---|---|
| PhCH3 | DMCb | n-BuOH | n-PrOH | EtOH | MeOH | ||
| a Compounds. b Dimethyl carbonate. | |||||||
| 1 | 61.5 | 5.53 | 5.88 | 6.05 | 6.07 | 6.10 | 6.43 |
| 2 | 62.0 | 6.14 | 8.02 | 8.08 | 8.63 | 9.18 | 10.20 |
| 3 | 62.4 | 7.25 | 10.01 | 10.13 | 10.23 | 10.30 | 11.20 |
| 4 | 68.7 | 49.05 | 56.72 | 57.82 | 64.07 | 70.35 | 82.33 |
| 5 | 66.8 | 9.94 | 12.85 | 13.33 | 13.71 | 14.10 | 15.83 |
| 6 | 65.0 | 5.69 | 6.46 | 6.58 | 6.76 | 6.93 | 7.40 |
The fluorous partition coefficient of compounds 1–6 in the toluene–CF3C6F11 binary mixture increased from 5.54 to 49.06 by increasing the fluorine content from 61.5% to 68.7% and the same trend can be observed for all other solvents (Fig. 2).
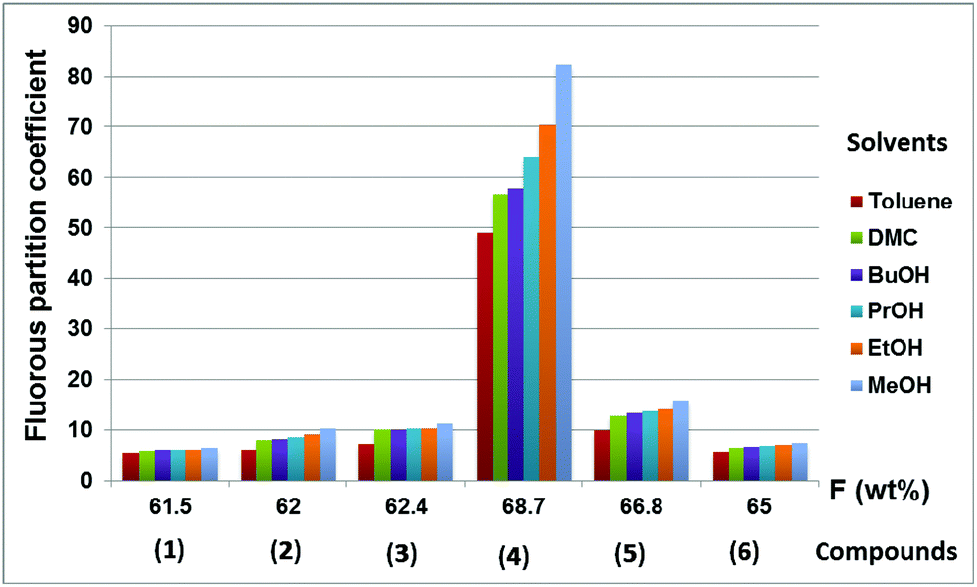 | ||
| Fig. 2 The fluorous partition coefficients (PF) of compounds 1–6 in different biphasic systems comprised of perfluoro(methylcyclohexane) and organic solvents. | ||
Of course, the fluorous partition coefficients were higher as the polarity of the organic solvents increased, reaching the maximum of 82.33 for 4 in the MeOH–CF3C6F11 biphasic system at room temperature. It should be noted that the increase of unsaturation in the hydrocarbon domain of compounds 1–3 resulted in slightly higher fluorous partition coefficients, indicating attractive interactions to form dimeric or oligomeric species.11c The important role of the overall fluorine content to achieve very high PF was clearly observable in the dramatic increase of PF from compound 5 to 4, suggesting again that the fluorous blanket should shield the hydrocarbon domain as much as possible to limit its interaction(s) with the other solvent.
The partition properties of biphasic systems can also be rationalized by the solvent properties of the liquids involved. Table 2 shows that the fluorophilicity (lnPF)7 of a fluorous compound increases when the organic solvent of the fluorous/organic biphasic systems has a higher Hildebrand solubility parameter (δ)15 and dielectric constant (ε)16 values.
P) of fluorous compounds 1–6a
| Solvent | δ | ε | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|---|---|
| a δ – Hildebrand parameter, ε – dielectric constant. | ||||||||
| PhCH3 | 8.91 | 2.4 | 1.711 | 1.815 | 1.981 | 3.893 | 2.297 | 1.738 |
| DMC | 9.92 | 3.2 | 1.772 | 2.082 | 2.304 | 4.038 | 2.553 | 1.866 |
| n-BuOH | 11.3 | 17.5 | 1.799 | 2.090 | 2.316 | 4.057 | 2.590 | 1.884 |
| n-PrOH | 11.97 | 203 | 1.804 | 2.155 | 2.326 | 4.160 | 2.618 | 1.912 |
| EtOH | 12.92 | 24.6 | 1.809 | 2.219 | 2.332 | 4.253 | 2.646 | 1.936 |
| MeOH | 14.28 | 32.7 | 1.862 | 2.323 | 2.416 | 4.411 | 2.762 | 2.001 |
The correlations between the fluorophilicity of the fluorous ethers 1–3 with the solvent properties of toluene and DMC were poor, but were excellent for alcohols (Fig. 3).
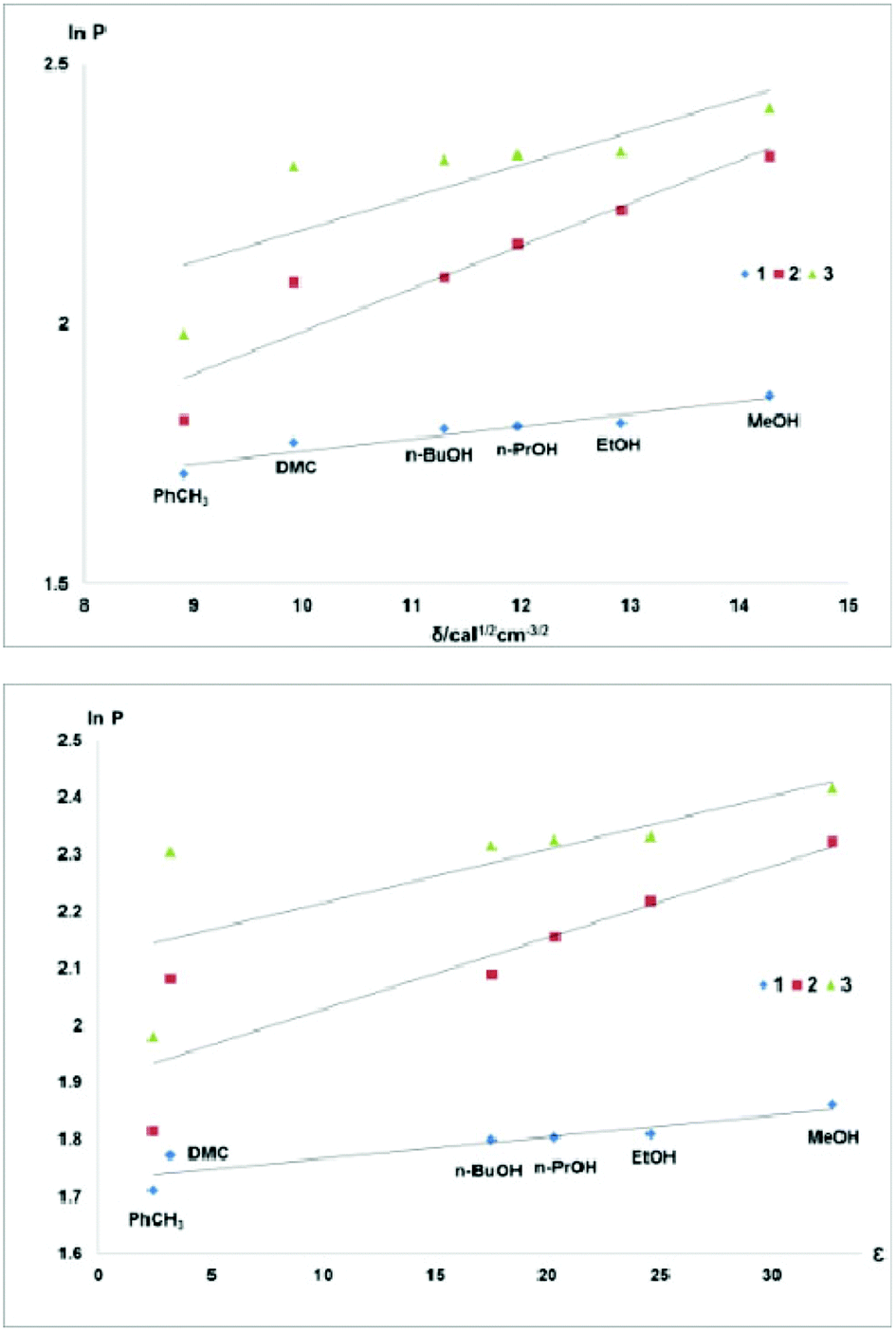 | ||
| Fig. 3 Fluorophilicity of compounds 1–3vs. the Hildebrand solubility parameter (top) or dielectric constant (bottom) of the solvents in biphasic systems comprised of perfluoro(methylcyclohexane) and different organic solvents. | ||
In contrast, the correlations between the fluorophilicity of the fluorous diethers 4–6 with the solvent properties of all solvents used were excellent (Fig. 4).
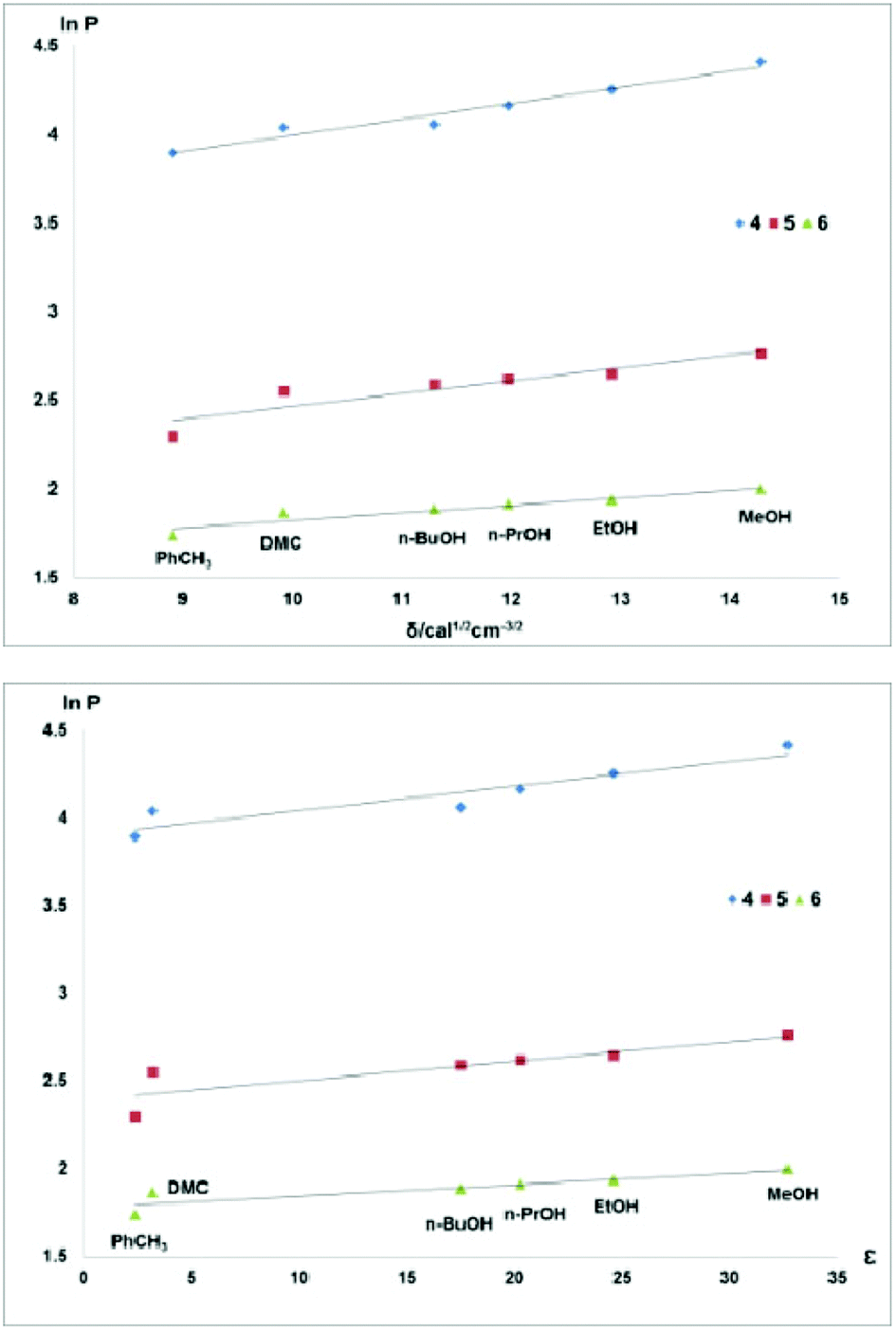 | ||
| Fig. 4 Fluorophilicity of compounds 4–6vs. the Hildebrand solubility parameter (top) or dielectric constant (bottom) of the solvents in biphasic systems comprised of perfluoro(methylcyclohexane) and different organic solvents. | ||
The toxicity of compounds 1–6 was also investigated using three human cell lines: A549 (lung carcinoma cells), HepG2 (hepatocellular carcinoma cells) and MDA-kb2 (mammary gland carcinoma cells). Nearly all of the fluorous compounds significantly reduced cell viability at the highest tested concentrations in all three cell lines. The A549 cells showed the greatest sensitivity, with four of the six tested compounds (1–4) significantly affecting the cell condition (Fig. 5).
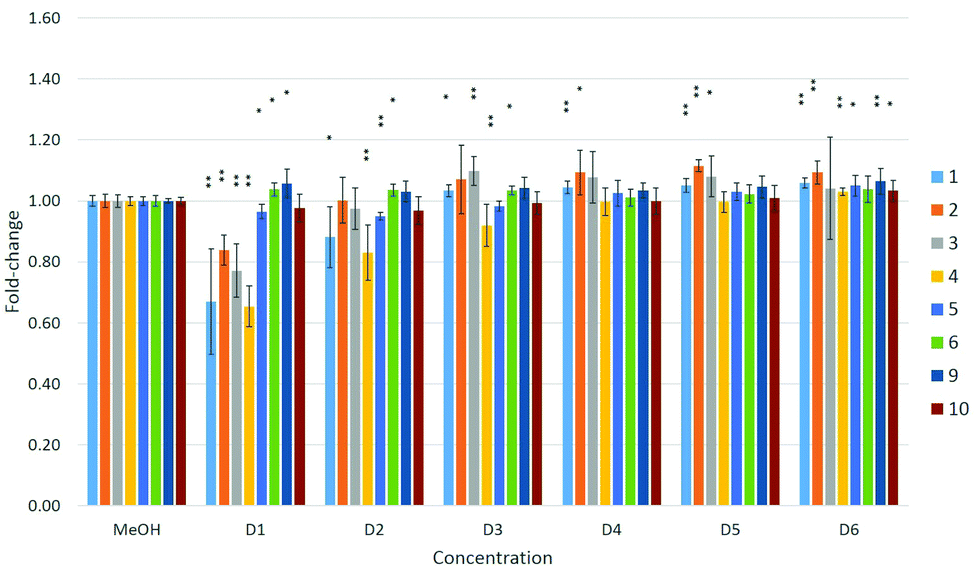 | ||
| Fig. 5 Effects of compounds 1–6 and 9–10 on cell viability in A549 cells. Effects are expressed as fold-changes in cell viability compared to MeOH controls. *p < 0.05, **p < 0.01; Mann–Whitney U. | ||
A dose–response relationship was observed for compound 4 at concentrations D1–D3 (see Table 4 in the Experimental section) in all three cell lines (Fig. 5–7), and for D1–D3 for 5 in MDA-kb2 cells (Fig. 7). In contrast, consistent and significant increases in cell viability were observed in MDA-kb2 at lower tested concentrations (D4–D6) for nearly all compounds, and similar increases were sporadically observed in HepG2 cells. In particular, 6 significantly increased cell viability at all tested concentrations in MDA-kb2 cells; the mean cell viability in these cases were all less than 1.5-fold of the solvent control viability.
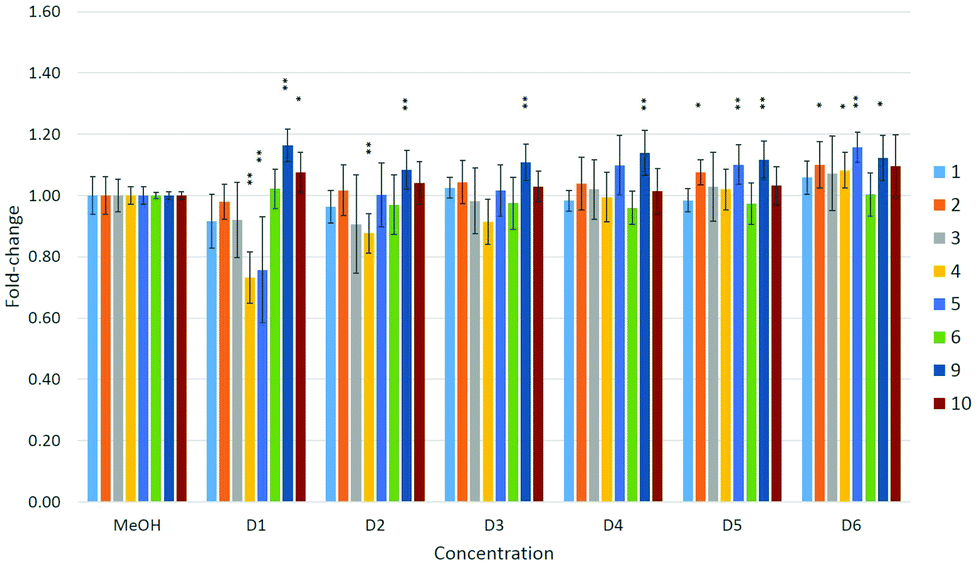 | ||
| Fig. 6 Effects of compounds 1–6 and 9–10 on cell viability in HepG2 cells. Effects are expressed as fold-changes in cell viability compared to MeOH controls. *p < 0.05, **p < 0.01; Mann–Whitney U. | ||
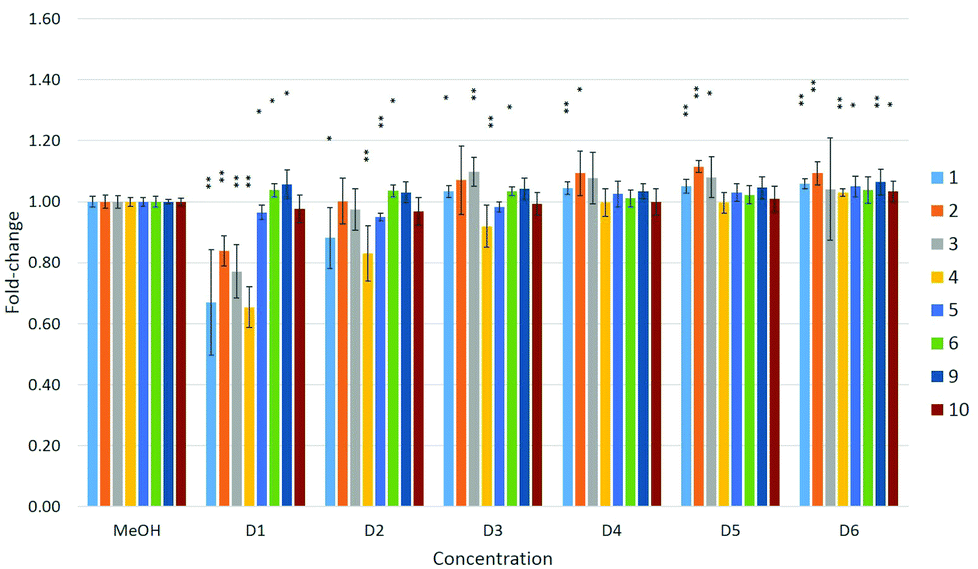 | ||
| Fig. 7 Effects of compounds 1–6 and 9–10 on cell viability in MDA-kb2 cells. Effects are expressed as fold-changes in cell viability compared to MeOH controls. *p < 0.05, **p < 0.01; Mann–Whitney U. | ||
Perfluorooctanoic acid (C7F15COOH, PFOA, 9) and sodium perfluoro-1-octanesulfonate (C8F17SO3Na, PFOS, 10) were also tested in the cells in parallel with the fluorous compounds to compare their cytotoxicities. Neither 9 nor 10 significantly reduced cell viability in any of the three cell lines at the tested concentrations. A previous study reported that 9 and 10 induced cytotoxicity in HepG2 cells after 24 hours of exposure at concentrations ≥200 μM,17 a concentration approximately 200 times greater than the highest concentration tested in this study (∼1 μM). Another investigation reported that C6–10 PFCAs induced cytotoxicity in promyelocytic leukemia rat IPC-81 cells and rat glioma C6 cells with EC50 values greater than approximately 170 μM; cytotoxicity was also found to increase with chain length.18 Similar results and a similar relationship between cytotoxicity and chain length were reported for C4 to C12 PFCAs tested in HepG2 cells, with EC50 values ranging from 7 μM to >1000 μM,19 and in human colon carcinoma HCT116 cells.20 In the present study, 4 was the most consistently cytotoxic of all the fluorous compounds, while 5 and 6 showed weaker effects on cell viability. The reason for the higher toxicity of 4 is not clear. One of the major mechanisms of action of 9 and 10 has been suggested to involve interactions with mammalian peroxisome proliferator-activated receptor α (PPARα),21 though the role of this mechanism in other reported effects, such as changes in development and immune function in mammals,22 is not understood. It is also possible that the greater toxicity of 4 is due to the formation of toxic metabolites in the cells during the exposure period; receptor interactions and the formation of potentially toxic metabolites will need to be investigated in future studies to assess these properties of the fluorous compounds. Nevertheless, it should be noted that, with the exception of 4, significant effects on cell viability were observed at concentrations approximately 3–30 times greater than those reported for 9 and 10.
Significant increases in cell viability were observed in HepG2 cells exposed to 9, and in MDA-kb2 cells exposed to 9 or 10, although the mean cell viability was less than 1.2-fold of solvent control levels. Cell proliferation of greater than two-fold of control levels as a result of exposure to 10 has been reported previously in HepG2 cells at concentrations between 5 and 50 μM, and the proliferative effect was observed to strengthen with chain length in cells exposed to PFCAs.4 The significant increases in cell viability observed in response to some of the fluorous compounds in the present study were not dose-dependent and were generally small; the reason for the consistent increases in viability after MDA-kb2 exposure to 6 is not clear.
The polymerization of F9-t-butyl allyl (2) and F9-t-butyl propargyl (3) ethers could be used to prepare polymers such as –{CH–C[CH2OC(CF3)3]}n– and –{CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH[CH2OC(CF3)3]}n–. We are currently focusing on the development of novel catalysts to polymerize 2 and 3. Although we have tested several known catalytic systems, so far we were unable to produce polymeric materials. This is probably due to the strong electron withdrawing effect of the F9-t-butoxy group resulting in very low reactivity.
CH[CH2OC(CF3)3]}n–. We are currently focusing on the development of novel catalysts to polymerize 2 and 3. Although we have tested several known catalytic systems, so far we were unable to produce polymeric materials. This is probably due to the strong electron withdrawing effect of the F9-t-butoxy group resulting in very low reactivity.
In order to gain insight into the partition equilibria of fluorous compounds in MeOH–CF3C6F11 and PhCH3–CF3C6F11 binary mixtures, density functional theory calculations were performed to predict structures and energetics of the fluorous diethers. A polarizable continuum model23 (PCM) was used to account for the solvent effect. The geometrical optimizations and vibrational frequencies calculations were carried out at the B3LYP level24 with the 6-31G(d) basis set in the presence of the solvent effect using Gaussian 09 program.25 Based on the mathematical relationship:
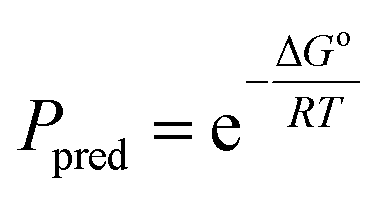 | (1) |
| Fluorous compounds and their oligomers | P F | P pred | |||
|---|---|---|---|---|---|
| PhCH3a |
MeOHb | PhCH3a |
MeOHb | ||
| a Biphasic system comprised of perfluoro(methylcyclohexane)–toluene. b Biphasic system comprised of perfluoro(methylcyclohexane)–methanol. | |||||
| 4 | Monomer | 49.05 | 82.33 | 1.66 | 70.83 |
| Dimer | 3.56 | 193.35 | |||
| Trimer | 6.81 | ||||
| Tetramer | 42.09 | ||||
| 5 | Monomer | 9.94 | 15.83 | 1.78 | 26.34 |
| Dimer | 3.76 | 416.53 | |||
| Trimer | 4.10 | ||||
| Tetramer | 8.29 | ||||
| 6 | Monomer | 5.69 | 7.40 | 1.75 | 16.33 |
| Dimer | 2.82 | 438.87 | |||
| Trimer | 5.28 | ||||
| Tetramer | 5.94 | ||||
The predicted partition coefficients of monomeric compounds 4, 5 and 6 in the MeOH–CF3C6F11 biphasic systems are 70.83, 26.34, and 16.33, respectively. These predictions are in a good agreement with the measured partition coefficients (see Table 1). In contrast, the respective partition coefficients (1.66, 1.78, and 1.75) for monomeric 4, 5 and 6 in PhCH3–CF3C6F11 biphasic systems are significantly smaller than the measured values. The partition coefficients are found to increase gradually from monomer to dimer to trimer to tetramer in PhCH3–CF3C6F11. The partition coefficients (42.09, 8.29 and 5.94) for the tetramer of 4, 5 and 6 are in good accord with the measured values (49.05, 9.94 and 5.69). The present theoretical predictions suggest that the fluorous diethers 4, 5 and 6 may exist in a monomeric form in MeOH–CF3C6F11, whereas they likely aggregate to form oligomers in PhCH3–CF3C6F11. The aggregation of the oligomers is stabilized by intermolecular hydrogen bond interactions (Fig. 8a–c).
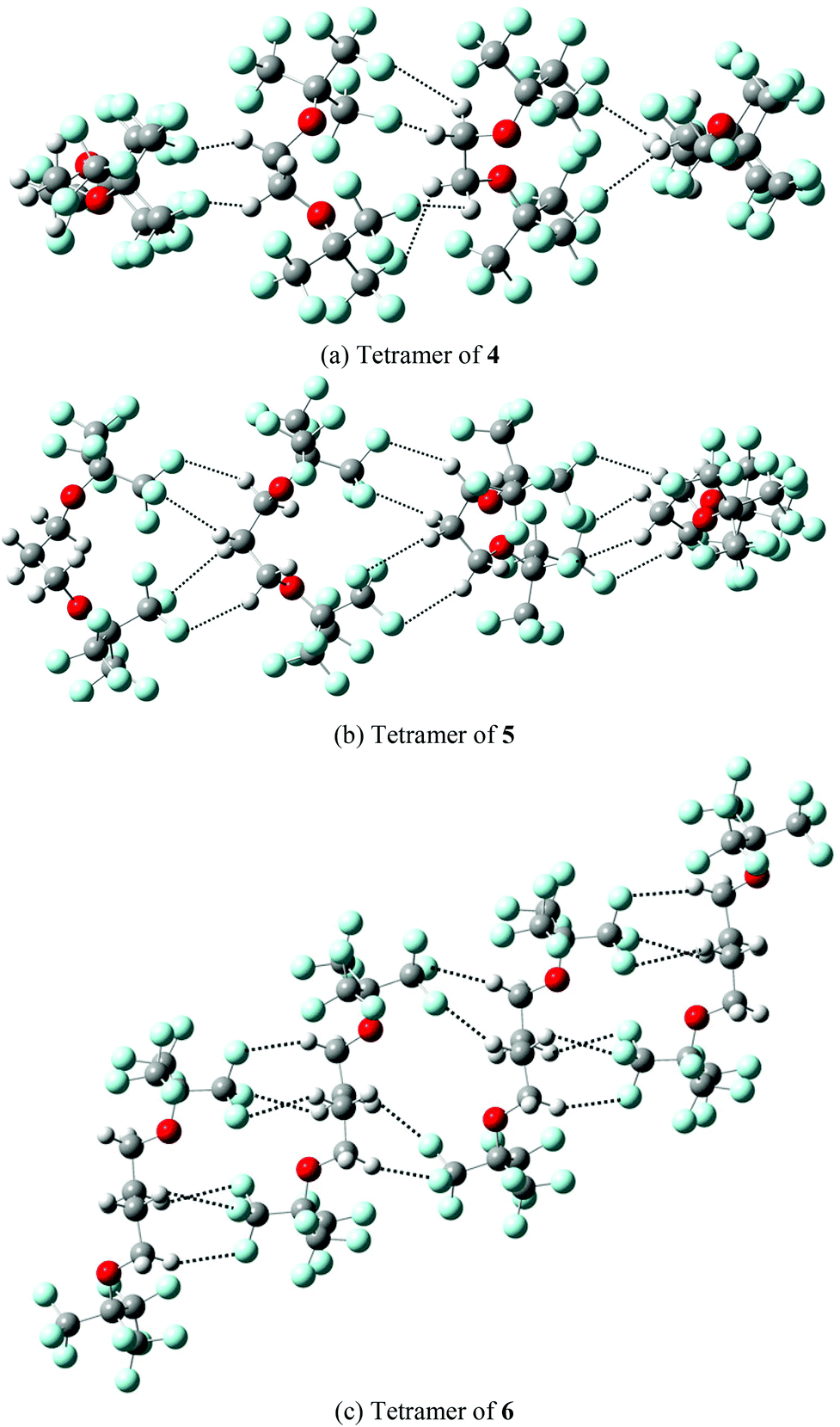 | ||
| Fig. 8 Optimized structures of (a) tetramer of 4; (b) tetramer of 5; (c) tetramer of 6 in the biphasic system comprised of perfluoro(methylcyclohexane) and toluene. The hydrogen bond interactions are shown in dotted lines. | ||
Experimental
Chemicals
Methanol, n-butanol, dimethyl carbonate, 1,2-dibromoethane, 1,3-dibromopropane, 1,4-dichlorobutane, n-propyl bromide, allyl bromide, and propargyl chloride were purchased from Aldrich. n-Propanol is the product of International Laboratory USA. Toluene, ethanol and N,N-dimethylformamide (DMF) are products of VWR. Perfluoro-tert-butanol and perfluoro(methyl-cyclohexane) are products of Fluorochem. Perfluorooctanoic acid or PFOA (9, purity >95%) Wellington Laboratories, Guelph, ON, Canada, and sodium perfluoro-1-octanesulfonate or PFOS (10, >99%) Apollo Scientific Ltd, Manchester, UK, were diluted in methanol for testing (Table 4). All purchased chemicals and solvents were used as received. Sodium perfloro-tert-butoxide was synthesized according to a literature method in 100% yield.261H-, 13C-, 19F-NMR spectra were recorded on a Bruker AV400 FT-NMR spectrometer at room temperature (see the NMR spectra in ESI†). Data were expressed as chemical shifts in ppm, relative to residual chloroform (1H-NMR δ = 7.26 ppm, 13C-NMR δ = 77.2 ppm), d6-acetone (1H-NMR δ = 2.05 ppm, 13C-NMR δ = 29.84 ppm) or an internal standard for 19F (hexafluorobenzene δ = −164.9 ppm). GC-MS were recorded on a Hewlett Packard 6890 GC instrument coupled with a 5973 mass selective detector. Infrared spectra were recorded in the range of 500–4000 cm−1 using a Nujol matrix or KBr plates on a Perkin-Elmer Model FTIR-1600 spectrometer.| Cpd | Stock [mM] | D1 [mM] | D2 [mM] | D3 [μM] | D4 [μM] | D5 [nM] | D6 [nM] |
|---|---|---|---|---|---|---|---|
| 1 | 46.42 | 9.28 | 0.928 | 92.84 | 9.28 | 928.36 | 92.84 |
| 2 | 46.73 | 9.35 | 0.935 | 93.46 | 9.35 | 934.57 | 93.46 |
| 3 | 51.71 | 10.34 | 1.034 | 103.41 | 10.34 | 1034.13 | 103.41 |
| 4 | 29.73 | 5.95 | 0.59 | 59.47 | 5.95 | 594.66 | 59.47 |
| 5 | 31.29 | 6.26 | 0.626 | 62.57 | 6.26 | 625.73 | 62.57 |
| 6 | 0.19 | 0.04 | 0.004 | 0.38 | 0.04 | 3.76 | 0.38 |
| 9 | 0.00095 | 0.00095 | 0.00019 | 0.019 | 0.002 | 0.190 | 0.019 |
| 10 | 0.0012 | 0.0012 | 0.00024 | 0.024 | 0.002 | 0.240 | 0.024 |
Measurement of the partition coefficients
A 7 mL vial was used to partition 0.010–0.030 mmol of 1–6 in a well-established biphasic system,27 which was prepared by mixing 2.000 mL of perfluoro(methylcyclohexane) and 2.000 mL of one of the organic solvents. The vial was sealed and vigorously mixed for several minutes. After standing at 25.0 °C for 24 hours, samples (0.500 mL) were taken from both phases, which were analysed by GC/MS using 1,4-dioxane as the internal standard. All partition experiments were done in triplicate.Toxicity measurements
All compounds were diluted in methanol (Sigma Aldrich, St. Louis, MO, USA) for toxicity testing. Stock solutions for the newly-synthesized fluorous compounds were prepared at concentrations ranging from 0.19–51.71 mM, and were diluted 1:5 and then serially diluted 1:10 to produce six concentrations for testing (Table 4).
Cell lines
Three human cell lines were chosen for toxicity testing: A549 (lung carcinoma cells), HepG2 (hepatocellular carcinoma cells) and MDA-kb2 (mammary gland carcinoma cells). All cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and were cultured in 75 cm2 culture flasks (NUNC, Thermo Scientific, Waltham, MA, USA). A549 and HepG2 cells were cultured in Kaighn's modification of Ham's F-12 medium (Sigma) and Dulbecco's Modified Eagle's/Ham's F-12 medium (Sigma), respectively, at 37 °C and 5% CO2. MDA-kb2 cells were cultured in L-15 medium (Gibco, Life Technologies, Grand Island, NY, USA) at 37 °C without supplemental CO2. All cell media were supplemented with 10% defined fetal bovine serum (Hyclone, Thermo Scientific).Cytotoxicity assays
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, which determines cytotoxicity by measuring the cellular mitochondrial reduction of the MTT dye to formazan crystals,28 was used to assess the cytotoxicities of the fluorous compounds. The assays were conducted for 5 days. Cells were seeded in 96-well plates (COSTAR, Corning, Tewksbury, MA, USA) at 250 μl of cell suspension per well on the first day; only the inner 60 wells of the plate were used to avoid edge effects, and blanks and methanol solvent control wells were included in each plate. A549 and HepG2 cells were seeded at densities of 2 × 105 and 7 × 104 cells ml−1, respectively, while MDA-kb2 cells were seeded at 1 × 105 cells ml−1. A549 and HepG2 cells were allowed to attach to 96-well plates for 24 h, while MDA-kb2 cells were dosed after 48 h; after attachment, all test chemicals were dosed at 1% v/v in triplicate. Wells were dosed individually. After dosing, the cells were incubated under culture conditions for 96 h (A549 and HepG2 cells) or 72 h (MDA-kb2 cells). After incubation, the MTT assays was conducted as described previously,28 and absorbance of the solubilized dye products was quantified using a SpectraMax M2e microplate reader (Molecular Devices, Sunnyvale, CA, USA). Cell assays were replicated twice for all cell lines.Statistical analysis
Cell viability was expressed as fold-change in terms of the relative absorbance (A) of the wells dosed with fluorous compounds compared to those of the solvent control:| Cell viability (fold-change) = (Asample/Asolvent control) |
Fold-change data were arcsine transformed and checked for homogeneity of variance and normality using SigmaPlot 11.0 (Systat Software Inc). Kruskal–Wallis tests followed by pairwise Mann–Whitney U post hoc tests were performed using SPSS version 17.0 (SPSS Inc., Chicago, IL) to compare differences among concentrations with respect to the methanol solvent controls. The data are expressed as means ± SD and were considered significant at p < 0.05.
Conclusions
Sodium nonafluoro-t-butoxide (7) was used as a convenient starting compound to synthesize fluorous ethers and diethers in good to high yields. Nonafluoro-t-butyl propyl (1), allyl (2), and propargyl (3) ethers as well as 1,2-bis(nonafluoro-t-butoxy)ethane (4), 1,3-bis(nonafluoro-t-butoxy)-propane (5), and 1,4-bis(nonafluoro-t-butoxy)-butane (6) were prepared and characterized. The fluorous partition coefficient of compounds 1–6 in the toluene–CF3C6F11 binary mixture increased significantly by increasing the fluorine content above 68% and was higher as the polarity of the organic solvents increased. Computational studies have shown that 4, 5 and 6 exist as a monomeric form in MeOH–CF3C6F11 and they likely aggregate to form oligomers in PhCH3–CF3C6F11. Some of the newly-synthesized fluorous compounds were cytotoxic to the tested cell lines, but with the exception of one compound, these effects were observed at concentrations approximately 3–30 times greater than those reported for widely used C8-perfluorinated compounds.Acknowledgements
We are grateful for the continuous support and help of the Technical Officers of the Department of Biology and Chemistry of City University of Hong Kong. This work was funded by the City University of Hong Kong, project number 7002604 (2010).Notes and references
- (a) A. E. C. Collis and I. T. Horváth, Catal. Sci. Technol., 2011, 1, 912 RSC; (b) Multiphase Homogeneous Catalysis, ed. B. Cornils, W. A. Herrmann, I. T. Horváth, W. Leitner, S. Mecking, H. Olivier-Bourbigou and D. Vogt, Wiley-VCH, Weinheim, 2005 Search PubMed.
- (a) I. T. Horváth and J. Rábai, Science, 1994, 266, 72 Search PubMed; (b) I. T. Horváth and J. Rábai, US5,463,082, 1995 Search PubMed.
- (a) A. F. M. Barton, Chem. Rev., 1975, 75, 731 CrossRef CAS; (b) A. F. M. Barton, CRC Handbook of Solubility Parameters and Other Cohesion Parameters, CRC Press, Inc, Boca Raton, Florida, 1983 Search PubMed.
- (a) Handbook of Fluorous Chemistry, ed. J. A. Gladysz, D. P. Curran and I. T. Horváth, Wiley-VCH, Weinheim, 2004 Search PubMed; (b) Fluorous Chemistry in Topics in Current Chemistry, ed. I. T. Horváth, Springer, Heidelberg, 2012 Search PubMed.
- R. B. Gupta and J.-J. Shim, Solubility in Supercritical Carbon Dioxide, CRC Press, Inc., Boca Raton, Florida, 2006 Search PubMed.
- V. Herrera, P. J. F. de Rege, I. T. Horváth, T. L. Husebo and R. P. Hughes, Inorg. Chem. Commun., 1998, 1, 197 CrossRef CAS.
- L. E. Kiss, I. Kovesdi and J. Rabai, J. Fluorine Chem., 2001, 108, 95 CrossRef CAS.
- (a) D. J. Luebker, K. J. Hansen, N. M. Bass, J. L. Butenhoff and A. M. Seacat, Toxicology, 2002, 176, 175 CrossRef CAS; (b) Provisional Health Advisories for Perfluorooctanoic Acid (PFOA) and Perfluorooctane Sulfonate (PFOS), US EPA, January 8, 2009 (http://www.epa.gov).
- I. T. Horváth, Changing Designer Issues in Fluorous Chemistry, 1st International Symposium on Fluorous Technologies, July 3–6, 2005, Bordeaux-Talence, France Search PubMed.
- (a) N. Kudo, N. Bandi, E. Suzuki, M. Katakura and Y. Kawashima, Chem.-Biol. Interact., 2000, 124, 119 CrossRef CAS; (b) K. P. Das, B. E. Grey, R. D. Zehr, C. R. Wood, J. L. Butenhoff, S.-C. Chang, D. J. Ehresman, Y.-M. Tan and C. Lau, Toxicol. Sci., 2008, 105, 173 CrossRef CAS PubMed.
- (a) A.-M. Bálint, A. Bodor, Á. Gömöry, K. Vékey, D. Szabó and J. Rábai, J. Fluorine Chem., 2005, 126, 1524 CrossRef PubMed; (b) A. Nemes, L. Tölgyesi, A. Bodor, J. Rábai and D. Szabó, J. Fluorine Chem., 2010, 131, 1368 CrossRef CAS PubMed; (c) X. Zhao, W. Y. Ng, K.-C. Lau, A. E. C. Collis and I. T. Horváth, Phys. Chem. Chem. Phys., 2012, 14, 3909 RSC; (d) T. Csóka, A. Nemes and D. Szabó, Tetrahedron Lett., 2013, 54, 1730 CrossRef PubMed.
- Q. Chu, C. Henry and D. P. Curran, Org. Lett., 2008, 10, 2453 CrossRef CAS PubMed.
- I. T. Horváth, K. C. Lau, M. Y. Lui, C. K. E. Law and K. C. P. Wong, US Patent Appl, 61/816,432, 2013 Search PubMed.
- R. Guterman, B. M. Berven, T. C. Corkery, H.-Y. Nie, M. Idacavage, E. R. Gillies and P. J. Ragogna, J. Poly. Sci., Part A: Polym. Chem., 2013, 51, 2782 CrossRef CAS.
- (a) J. Hildebrand and R. L. Scott, The Solubility of Nonelectrolytes, Reinhold, New York, 3rd edn, 1950 Search PubMed; (b) J. Hildebrand and R. L. Scott, Regular Solutions, Prentice-Hall Inc., Englewood Cliffs, NJ, 1962 Search PubMed.
- P. Muller, Pure Appl. Chem., 1994, 66, 1077 CrossRef.
- A. Florentin, T. Deblonde, N. Diguio, A. Hautemaniere and P. Harteman, Int. J. Hyg. Environ. Health, 2011, 214, 493 CrossRef CAS PubMed.
- E. Mulkiewicz, B. Jastorff, A. C. Składanowski, K. Kleszczyński and P. Stepnowski, Environ. Toxicol. Pharmacol., 2007, 23, 279 CrossRef CAS PubMed.
- T. Buhrke, A. Kibellus and A. Lampen, Toxicol. Lett., 2013, 218, 97 CrossRef CAS PubMed.
- K. Kleszczyński, P. Gardzielewski, E. Mulkiewicz, P. Stepnowski and A. C. Składanowski, Toxicol. In Vitro, 2007, 21, 1206 CrossRef PubMed.
- M. L. Takacs and B. D. Abbott, Toxicol. Sci., 2007, 95, 108 CrossRef CAS PubMed.
- A. B. Lindstrom, M. J. Strynar and E. L. Libelo, Environ. Sci. Technol., 2011, 45, 7954 CrossRef CAS PubMed.
- (a) S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef CAS; (b) S. Miertus and J. Tomasi, Chem. Phys., 1982, 65, 239 CrossRef CAS.
- A. D. Beck, J. Chem. Phys., 1993, 98, 5648 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision B.1), Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- D. Szabó, J. Mohl, A.-M. Bálint, A. Bodor and J. Rábai, J. Fluorine Chem., 2006, 127, 1496 CrossRef PubMed.
- J. A. Gladysz, C. Emnet and J. Rábai, Partition Coefficients Involving Fluorous Solvents, in Handbook of Fluorous Chemistry, ed. J. A. Gladysz, D. P. Curran and I. T. Horváth, Wiley-VCH, Weinheim, 2004, ch. 6 Search PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 16, 55 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C4QO00192C |
| This journal is © the Partner Organisations 2014 |