
Highly efficient and stereocontrolled oxidative coupling of tetrahydropyrroloindoles: synthesis of chimonanthines, (+)-WIN 64821 and (+)-WIN 64745†
Deqian
Sun‡
b,
Changyu
Xing‡
a,
Xiaoqing
Wang
a,
Zhongquan
Su
a and
Chaozhong
Li
*ab
aShanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
bDepartment of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: clig@mail.sioc.ac.cn
First published on 25th July 2014
Abstract
The highly efficient and stereocontrolled dimerization of 1,2,3,8-tetrahydropyrrolo[2,3-b]indoles was successfully developed with I2 as the oxidant, which allowed the rapid synthesis of meso- and rac-chimonanthines, (+)-WIN 64821 and (+)-WIN 64745 in the highest overall yields to date via the oxidation–oxidation–reduction sequence.
Dimeric hexahydropyrrolo[2,3-b]indole alkaloids are a unique class of natural bisindole alkaloids exhibiting a diverse range of biological activities.1 In particular, the dimeric pyrroloindole alkaloids with the C(3a)–C(3a′) linkage possess the fascinating architecture of vicinal quaternary stereocenters adjacent to two aminals. Typical examples are meso-chimonanthine (1) and rac-chimonanthine (2), as well as the dimeric diketopiperazine alkaloids (+)-WIN 64821 (3) and (+)-WIN 64745 (4),2 as shown in Fig. 1. These complex structural features have attracted considerable attention and their construction in a stereocontrolled manner continues to be actively pursued.3–12
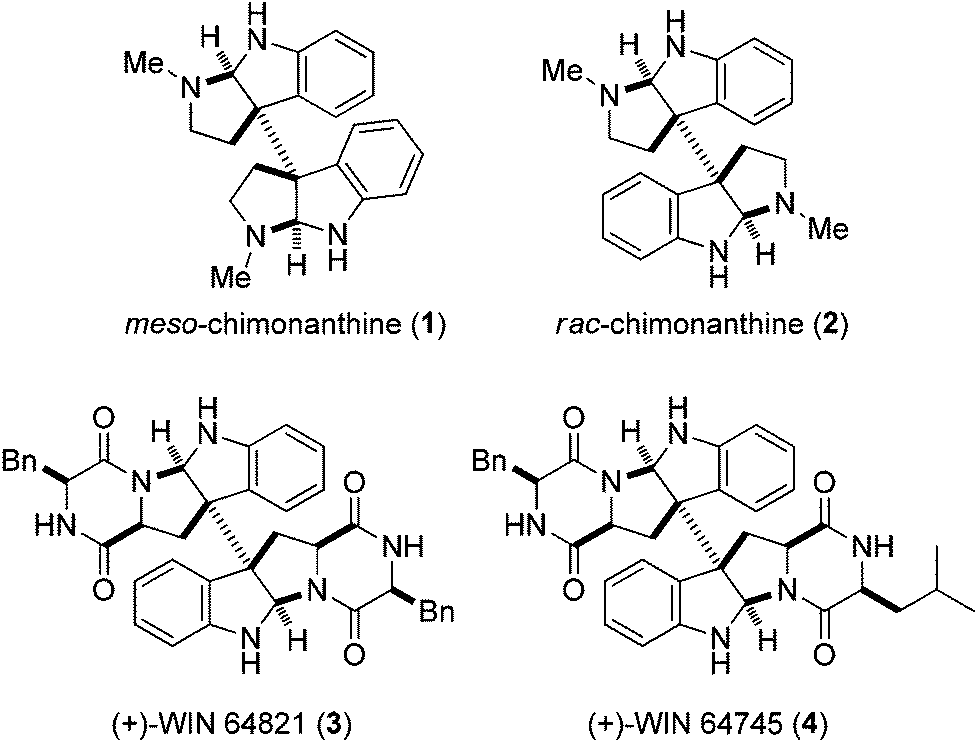 | ||
| Fig. 1 Representative examples of dimeric hexahydropyrroloindole alkaloids. | ||
A number of methods have been developed for the synthesis of these complex molecules, which generally fall into three major categories as depicted in Fig. 2. In the first category, dihydroisoindigo (A) derivatives were used as the starting materials or served as synthetic intermediates.4,5 This was exemplified by Overman's first enantioselective total synthesis of chimonanthines.4b The second category, pioneered by Movassaghi et al.,6 involved the Co(I)-mediated reductive dimerization of 8-protected 3a-bromohexahydropyrroloindoles such as compound B.6–8 This was showcased by the concise synthesis of 3, 4 and the more complexed ones such as (+)-11,11′-dideoxyverticillin A.6c Nevertheless, an excess amount of Co(I) complex was required and the yields of dimerization were not high (∼60%). The dimerization could also be mediated with Zn/Ni or Mn/Ni, but in low efficiency and poor stereoselectivity.9 The third approach was biomimetic oxidative coupling of tryptamine derivatives (C), providing a rapid access to the target molecules.10,11 However, it was disappointing to see that in all cases the yields were low and/or the stereoselectivity was poor. The development of more efficient and general methods is certainly highly desirable. Herein we report the facile synthesis of 1–4via I2-mediated, highly efficient and stereocontrolled oxidative dimerization of tetrahydropyrroloindoles (D).
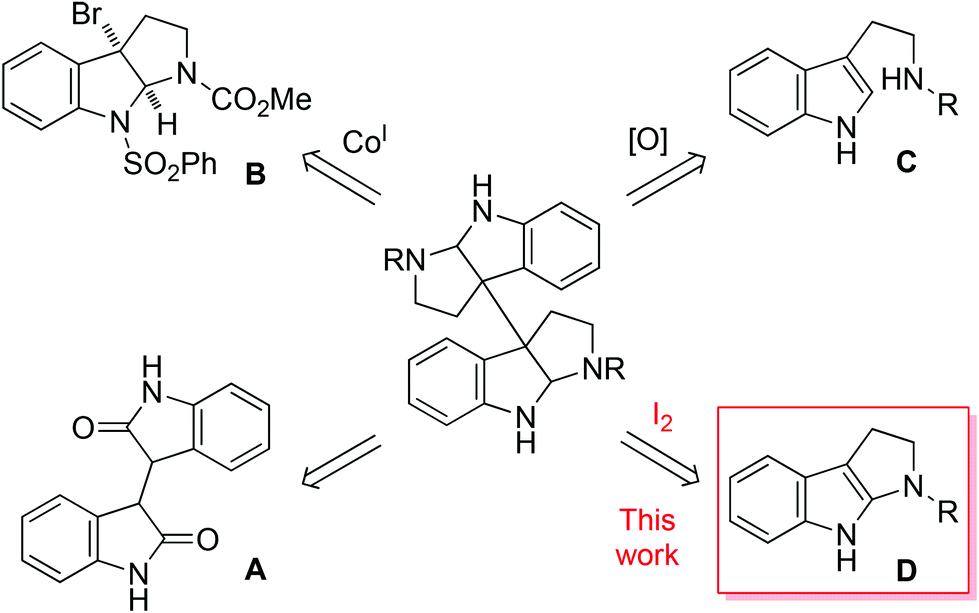 | ||
| Fig. 2 Representative synthetic approaches towards chimonanthines. | ||
Our idea originated from the biosynthetic hypotheses of chimonanthines. It has long been postulated by Robinson and Teuber13 that the biosynthesis of chimonanthine compounds involves the oxidative dimerization of two tryptamine units (E) via the benzyl radical intermediates F (Fig. 3). However, an alternative pathway has recently been proposed by Movassaghi et al.,3c which involves the repeated oxidation and rearrangement of bisindole G to produce bisamidine H followed by subsequent reduction. Encouraged by Movassaghi's perception, we suggest herein the third route as the hybrid of the above two hypotheses: tryptamine E rather than bisindole G might undergo the oxidation–oxidation–reduction sequence to produce chimonanthines via Movassaghi's intermediate bisamidine H (Fig. 3).
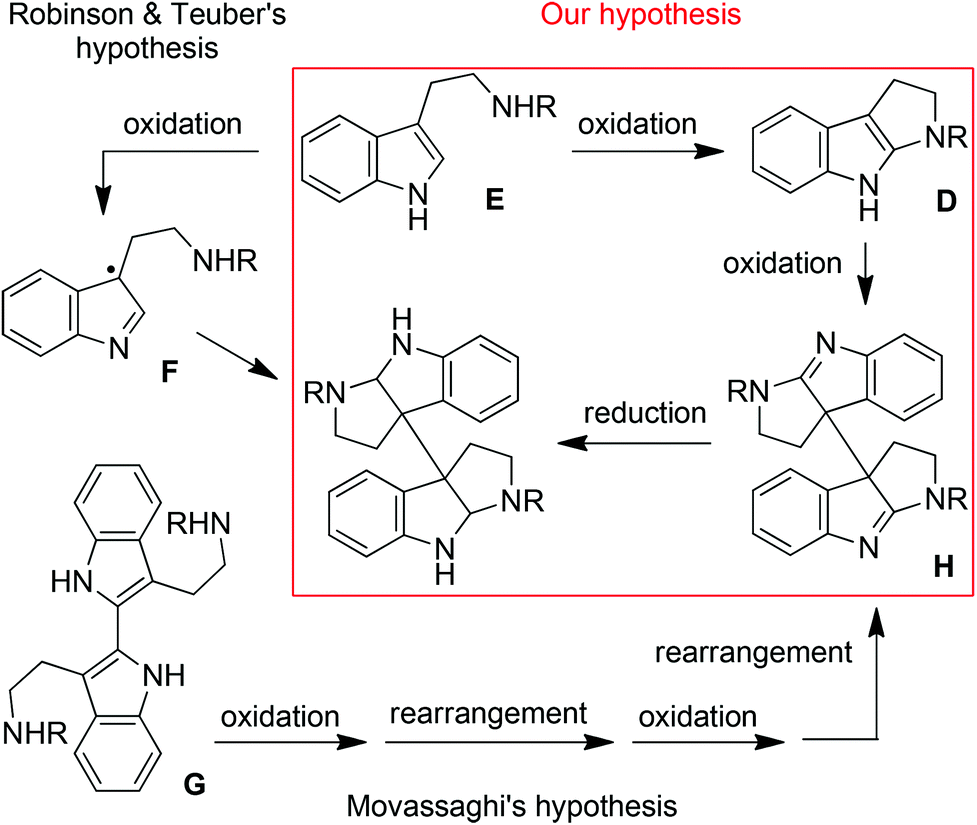 | ||
| Fig. 3 Hypotheses of the biosynthesis of chimonanthines. | ||
Thus, 1-nosyltetrahydropyrroloindole (5) was initially used to test our hypothesis. Substrate 5 was readily prepared in 78% yield from N-nosyl tryptamine by reaction with tBuOCl/Et3N according to the literature methods.14 The treatment of 5 with lithium bis(trimethylsilyl)amide (LiHMDS) and iodine15 in THF at 0 °C afforded the coupling products in 53% yield as the mixture of 6 and 7 in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, whose structures were unambiguously established by their X-ray diffraction experiments (Scheme 1).16 Interestingly, when NaH was used as the base, the reaction of 5 with I2 gave the mixture of 6 and 7 in a 1:10 ratio in a combined yield of 67%.
:1 ratio, whose structures were unambiguously established by their X-ray diffraction experiments (Scheme 1).16 Interestingly, when NaH was used as the base, the reaction of 5 with I2 gave the mixture of 6 and 7 in a 1:10 ratio in a combined yield of 67%.
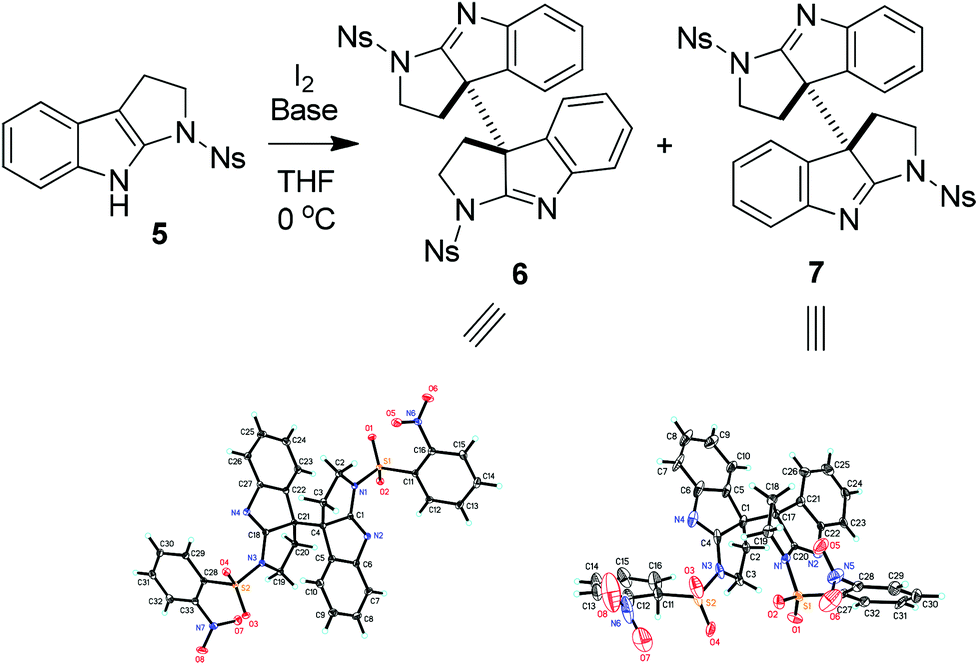 | ||
| Scheme 1 Oxidative coupling of 5. | ||
Intrigued by the above results, we decided to study the oxidative coupling in detail. 1-(Benzoxycarbonyl)-tetrahydropyrroloindole (8) rather than 5 was then chosen as the model in consideration of simplifying the total synthesis of 1 and 2 (vide infra). The results are summarized in Table 1. With NaH as the base and THF as the solvent, the oxidation of 8 using I2 at 0 °C for 10 min gave the coupling products 9 (17%) and 10 (72%). The efficiency and stereoselectivity were both increased when KH was used as the base (entries 1 and 2). Lowering the temperature increased the yield of 9 while the stereoselectivity was dramatically reversed when the solvent was switched to DMF (entries 3 and 4). On the other hand, the formation of 9 was partially inhibited when the reaction concentration was diluted from 0.1 M to 0.02 M (entries 2 and 5). KHMDS and NaHMDS showed similar effects to KH and NaH in terms of stereoselectivity (entries 6 and 7). However, with LiHMDS as the base, a high selectivity in favor of 9 was now observed. When the temperature was lowered to −40 °C, compound 9 was obtained in 95% yield almost exclusively (entries 8 and 9). Note that in all cases the oxidative dimerization of 8 showed an excellent efficiency, in sharp contrast to the direct oxidative coupling of its tryptamine precursors. The dramatically different effects of alkali metal ions on the stereoselectivity of dimerization were also remarkable.
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Base | Solvent | Temp (°C) | Yieldb (%) | |
| 9 | 10 | ||||
| a Reaction conditions: 8 (0.20 mmol), I2 (0.15 mmol), base (0.22 mmol), solvent (2 mL), 10 min. b Isolated yield based on 8. c THF (10 mL) was used. | |||||
| 1 | NaH | THF | 0 | 17 | 72 |
| 2 | KH | THF | 0 | 14 | 85 |
| 3 | KH | DMF | 0 | 66 | 33 |
| 4 | KH | THF | −40 | 21 | 69 |
| 5c | KH | THF | 0 | 7 | 87 |
| 6 | KHMDS | THF | 0 | 14 | 76 |
| 7 | NaHMDS | THF | 0 | 22 | 62 |
| 8 | LiHMDS | THF | 0 | 80 | 10 |
| 9 | LiHMDS | THF | −40 | 95 | Trace |
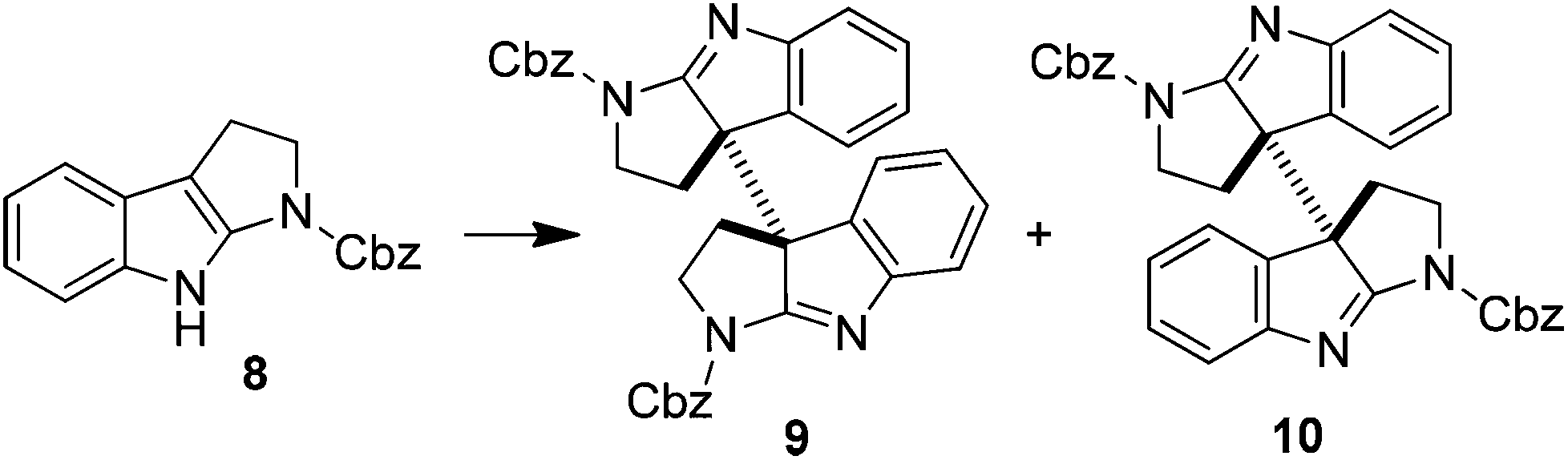
The synthesis of meso- and rac-chimonanthines thus becomes simple based on the above results. The four-step stereocontrolled synthesis of 1 starting from the readily available tryptamine 11 is summarized in Scheme 2. Compound 9 was reduced to 12 with catecholborane (CatBH) at RT. The subsequent conversion of the cbz groups to methyl ones with Red-Al furnished meso-chimonanthine 1 in an overall 38% yield. In a similar fashion (Scheme 3), the reduction of 10 with NaBH3CN gave compound 13, which was readily transformed to 2 with Red-Al in an overall 40% yield (based on 11). The spectra of 1 and 2 thus synthesized were identical to those reported in the literature.4a,12a
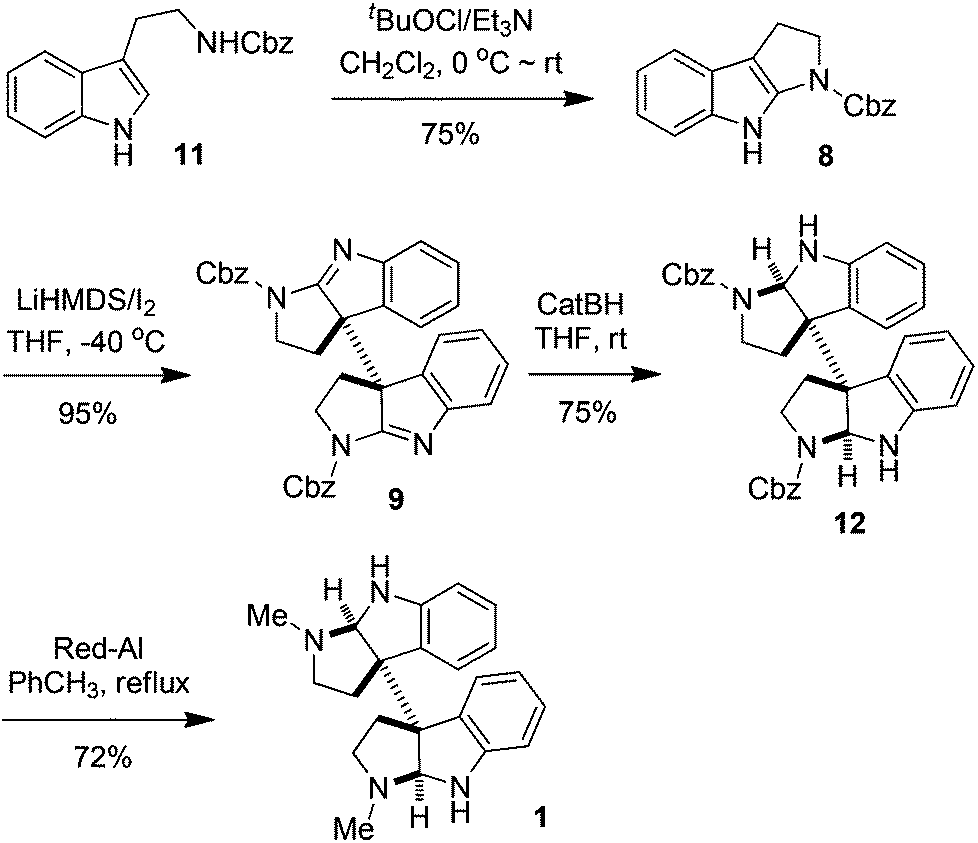 | ||
| Scheme 2 Synthesis of meso-chimonanthine. | ||
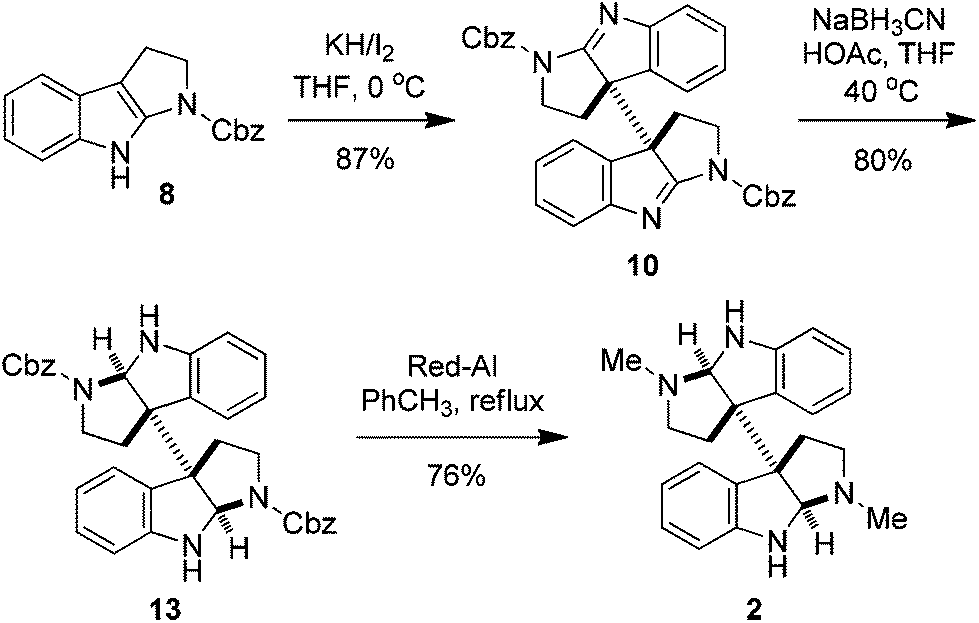 | ||
| Scheme 3 Synthesis of rac-chimonanthine. | ||
The above results also prompted us to extend the oxidation–oxidation–reduction sequence to tryptophan derivatives. Thus the oxidation of N-Cbz-L-tryptophan methyl ester (14) with tBuOCl/Et3N gave tetrahydropyrroloindole 15 (83% yield). Further oxidative dimerization of 15 with NaH/I2 in THF at 0 °C produced bisamidine 16 in 80% yield as a single diastereoisomer, whose structure was firmly established by its X-ray diffraction experiments17 (see the ESI†).18 The NaBH3CN reduction of 16 (88% yield) followed by deprotection with Pd/C–H2 (95% yield) led to compound 17. The O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU)19/Et3N-promoted condensation of 17 with Fmoc-L-phenylalanine followed by the removal of the Fmoc group by Et2NH at RT furnished 3 in 65% yield (Scheme 4).20 Thus (+)-WIN 64821 was synthesized in six steps starting from 14 in an overall 36% yield.
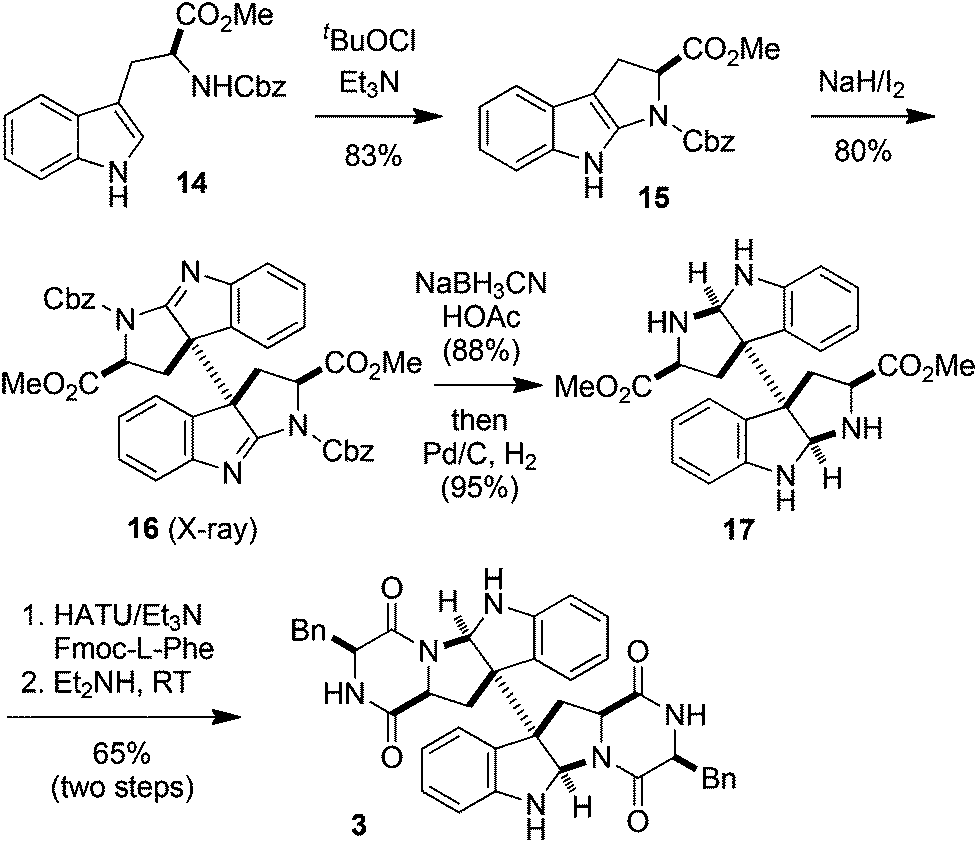 | ||
| Scheme 4 Synthesis of (+)-WIN 64821. | ||
The intermediate 17 could also be utilized for the synthesis of (+)-WIN 64745. As shown in eqn (1), the successive condensation of 17 with Fmoc-L-leucine and Fmoc-L-phenylalanine followed by deprotection with Et2NH gave rise to the target molecule 4 in 54% yield. This constitutes the six-step synthesis of (+)-WIN 64745 starting from tryptophan 14 with an overall 30% yield. The spectra of 3 and 4 thus synthesized were identical with those reported in the literature.7a Furthermore, it is conceivable that optically active (+)-chimonanthine (+)-2 can also be synthesized from 17via N-methylation and decarboxylation.21
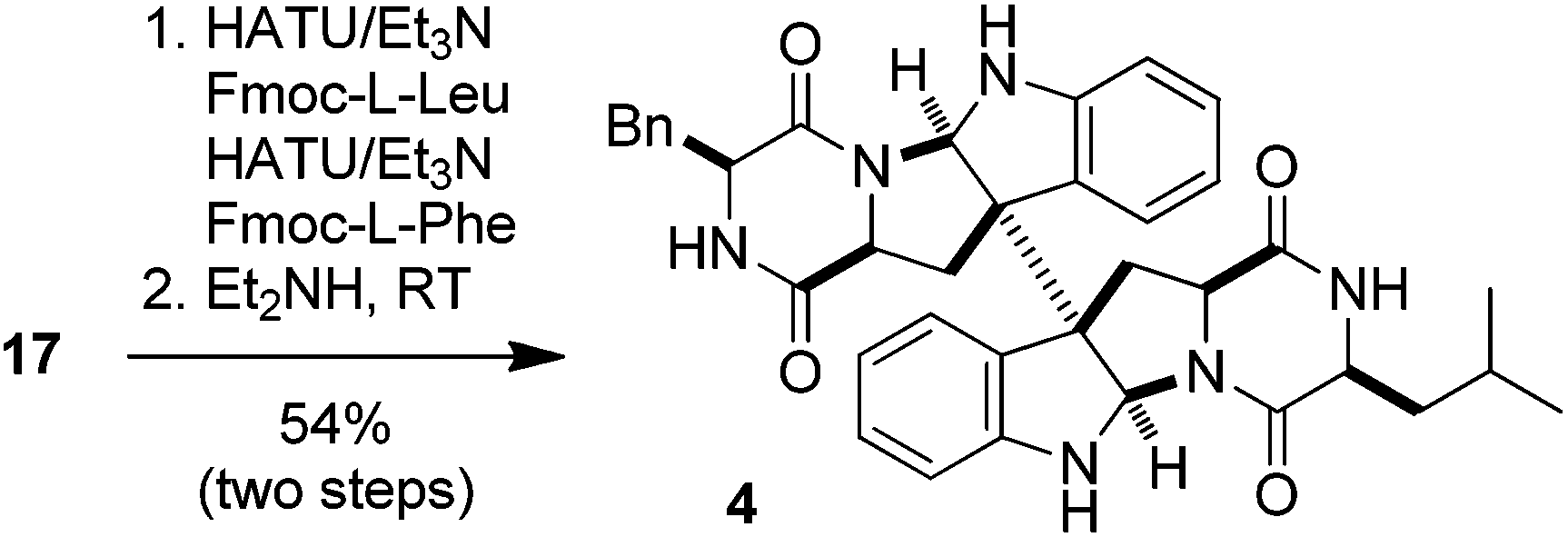 | (1) |
To gain more insight into the highly efficient and stereoselective dimerization of tetrahydropyrroloindoles, we designed the following experiments. In the presence of 10 equivalents of 2,2,6,6-tetramethylpiperidin-1-oxy radical (TEMPO), the treatment of 8 with LiHMDS/I2 at −40 °C gave the products 9 (93% yield) and 10 (5% yield), indicating that TEMPO had no effect on the dimerization. TEMPO showed no influence at all on the reaction of 8 with KH/I2 either. These two experiments indicate that the dimerization of 8 is unlikely a free radical process. Next, N-bromosuccinimide (NBS) was used as the substitute for iodine. The reaction of 5 with LiHMDS/NBS at −78 °C led to the formation of bromide 18 in 83% yield. Treatment of 5 with equimolar amount of LiHMDS followed by the addition of bromide 18 at RT furnished the mixture of 6 and 7 in 47% yield (eqn (2)). Finally, 1,2,3,4-tetrahydrocyclopenta[b]indole (19) was prepared as the substitute for tetrahydropyrroloindoles 5 and 8. However, the reaction of 19 with LiHMDS/I2 or KH/I2 gave no expected dimerization product at all and most of the starting material 19 was recovered after the usual workup (eqn (3)).
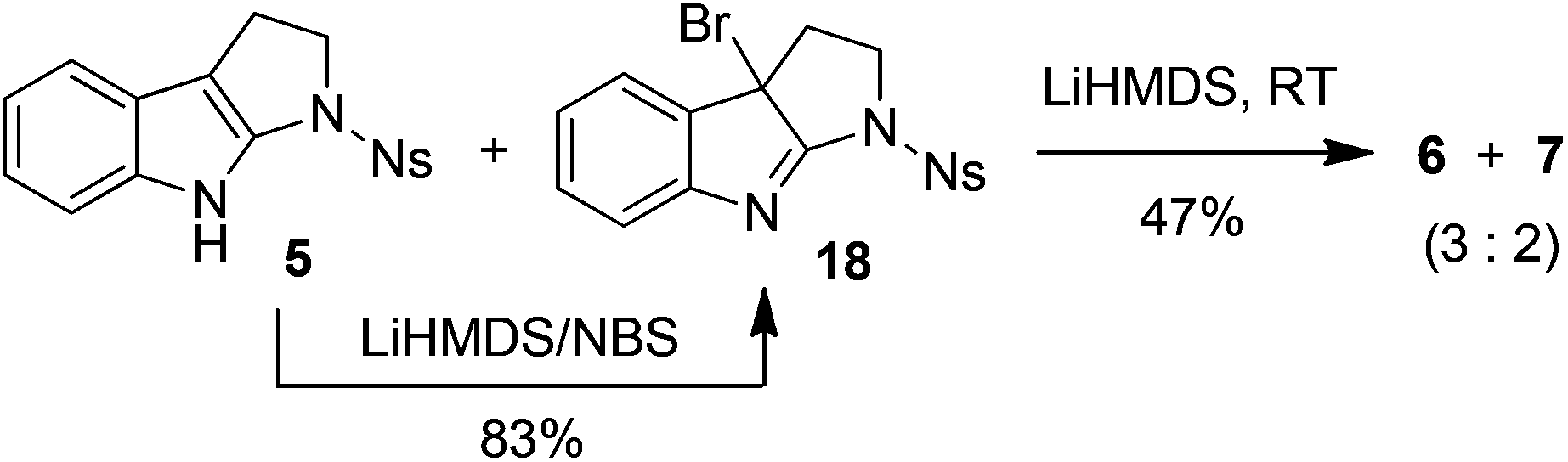 | (2) |
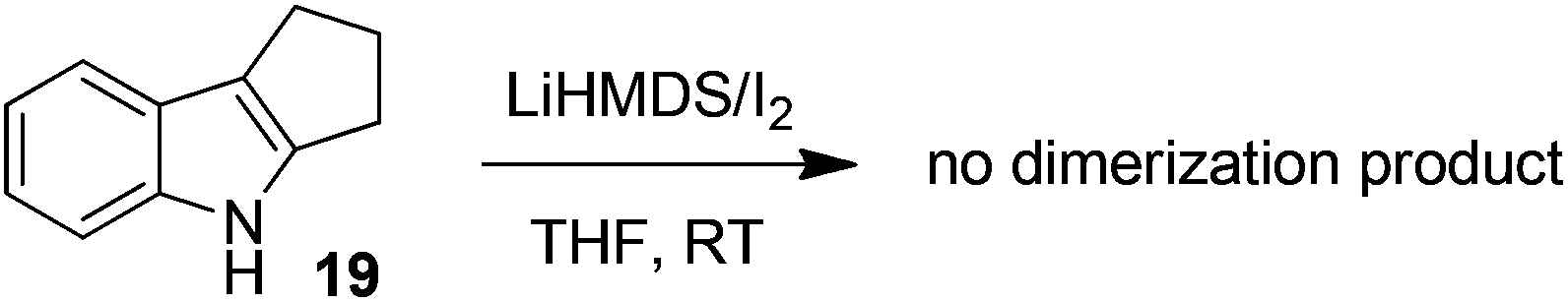 | (3) |
A plausible mechanism is therefore proposed based on the above mechanistic studies (Fig. 4). The deprotonation of 8 by a strong base generates the anion H or its tautomer I, which affords the iodide J on reaction with iodine. The intermediate J then loses an iodide ion to give the carbocation L presumably driven by the nucleophilicity of the amide nitrogen. The coupling between K and I led to the dimerization products 9 and/or 10. Note that only a half equivalent of I2 is required according to this mechanism, consistent with our experimental results in Table 1. The failure of 19 in dimerization can thus be attributed to the lack of driving force in the iodide elimination step (from J to L). Furthermore, the stereoselectivity of dimerization shown in Table 1 might be rationalized by M1 or M2 intermediacy shown in Fig. 4. In the case of LiHMDS as the base, the lithium ion chelates to the carbonyl oxygen and the nitrogen atom. The coupling between K and I then adopts the conformations of least steric hindrance such as M1 to give bisamidine 9. On the other hand, in the case of KH as the base, the potassium ion coordinates to two carbonyl oxygen atoms in K and I, and thus the coupling proceeds viaM2-like intermediacy to give 10. The use of DMF as solvent destabilizes M2 and therefore lowers the stereoselectivity, in excellent agreement with the experimental observation (entry 3, Table 1). The above mechanistic discussion might also shed light on the mechanisms of other I2-mediated oxidative coupling reactions.15
 | ||
| Fig. 4 Proposed mechanism for the dimerization of 8. | ||
In conclusion, we have successfully developed a new strategy, the oxidation–oxidation–reduction sequence, for the construction of the 3a,3a′-bis(hexahydropyrrolo[2,3-b]indole) skeleton based on our hypothesis of the biosynthesis of chimonanthines. Our approach features the I2-mediated, highly efficient oxidative dimerization of 1,2,3,8-tetrahydropyrrolo[2,3-b]indoles in a stereocontrolled manner, thus enabling the convenient synthesis of meso- and rac-chimonanthines, (+)-WIN 64821 and (+)-WIN 64745 in the highest overall yields to date. The key role of 1,2,3,8-tetraydropyrrolo-indoles demonstrated above also sheds light on the more challenging synthesis22 of higher-order hexahydropyrroloindole alkaloids (such as hodgkinsine and quadrigemine C)3 having the 3a,7′-connection, which is actively pursued in our laboratory.
Acknowledgements
This project was supported by the National Natural Science Foundation of China (grant no. 21228202, 21272259, 21290180 and 21361140377) and by the National Basic Research Program of China (973 Program) (grant no. 2011CB710805).References
- (a) G. A. Cordell and J. E. Saxton, Alkaloids, 1981, 20, 1–295 CAS; (b) T. Hino and M. Nakagawa, Alkaloids, 1988, 34, 1–75 CAS; (c) S. Hibino and T. Choshi, Nat. Prod. Rep., 2001, 18, 66–87 RSC; (d) M. Ishikura and K. Yamada, Nat. Prod. Rep., 2009, 26, 803–852 RSC.
- C. J. Barrow, P. Cai, J. K. Snyder, D. M. Sedlock, H. H. Sun and R. Cooper, J. Org. Chem., 1993, 58, 6016–6021 CrossRef CAS.
- For reviews, see: (a) D. Crich and A. Banerjee, Acc. Chem. Res., 2007, 40, 151–161 CrossRef CAS PubMed; (b) A. Steven and L. E. Overman, Angew. Chem., Int. Ed., 2007, 46, 5488–5508 CrossRef CAS PubMed; (c) M. A. Schmidt and M. Movassaghi, Synlett, 2008, 313–324 CAS; (d) J. Kim and M. Movassaghi, Chem. Soc. Rev., 2009, 38, 3035–3050 RSC; (e) S. Tadano and H. Ishikawa, Synlett, 2014, 157–162 Search PubMed; (f) P. Ruiz-Sanchis, S. A. Savina, F. Albericio and M. Alvarez, Chem. – Eur. J., 2011, 17, 1388–1408 CrossRef CAS PubMed.
- (a) J. T. Link and L. E. Overman, J. Am. Chem. Soc., 1996, 118, 8166–8167 CrossRef CAS; (b) L. E. Overman, D. V. Paone and B. A. Stearns, J. Am. Chem. Soc., 1999, 121, 7702–7703 CrossRef CAS; (c) L. E. Overman, J. F. Larrow, B. A. Stearns and J. M. Vance, Angew. Chem., Int. Ed., 2000, 39, 213–215 CrossRef CAS; (d) L. E. Overman and D. V. Paone, J. Am. Chem. Soc., 2001, 123, 9465–9467 CrossRef CAS.
- (a) S. Ghosh, S. Bhunia, B. N. Kakde, S. De and A. Bisai, Chem. Commun., 2014, 50, 2434–2437 RSC; (b) B. M. Trost and M. Osipov, Angew. Chem., Int. Ed., 2013, 52, 9176–9181 CrossRef CAS PubMed; (c) C. Guo, J. Song, J.-Z. Huang, P.-H. Chen, S.-W. Luo and L.-Z. Gong, Angew. Chem., Int. Ed., 2012, 51, 1046–1050 CrossRef CAS PubMed; (d) H. Mitsunuma, M. Shibasaki, M. Kanai and S. Matsunaga, Angew. Chem., Int. Ed., 2012, 51, 5217–5221 CrossRef CAS PubMed; (e) C.-L. Fang, S. Horne, N. Taylor and R. Rodrigo, J. Am. Chem. Soc., 1994, 116, 9480–9486 CrossRef CAS; (f) J. B. Hendrickson, R. Göschke and R. Rees, Tetrahedron, 1964, 20, 565–579 CrossRef CAS; (g) T. Hino and S.-i. Yamada, Tetrahedron Lett., 1963, 4, 1757–1760 CrossRef.
- (a) M. Movassaghi and M. A. Schmidt, Angew. Chem., Int. Ed., 2007, 46, 3725–3728 CrossRef CAS PubMed; (b) M. Movassaghi, M. A. Schmidt and J. A. Ashenhurst, Angew. Chem., Int. Ed., 2008, 47, 1485–1487 CrossRef CAS PubMed; (c) J. Kim, J. A. Ashenhurst and M. Movassaghi, Science, 2009, 324, 238–241 CrossRef CAS PubMed; (d) J. Kim and M. Movassaghi, J. Am. Chem. Soc., 2010, 132, 14376–14378 CrossRef CAS PubMed.
- (a) C. Pérez-Balado and Á. R. de Lera, Org. Lett., 2008, 10, 3701–3704 CrossRef PubMed; (b) C. Pérez-Balado, P. Rodríguez-Graña and Á. R. de Lera, Chem. – Eur. J., 2009, 15, 9928–9937 CrossRef PubMed; (c) W. Xie, G. Jiang, H. Liu, J. Hu, X. Pan, H. Zhang, X. Wan, Y. Lai and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 12924–12927 CrossRef CAS PubMed.
- E. Iwasa, Y. Hamashima, S. Fujishiro, E. Higuchi, A. Ito, M. Yoshida and M. Sodeoka, J. Am. Chem. Soc., 2010, 132, 4078–4079 CrossRef CAS PubMed.
- (a) Y. Peng, L. Luo, C.-S. Yan, J.-J. Zhang and Y.-W. Wang, J. Org. Chem., 2013, 78, 10960–10967 CrossRef CAS PubMed; (b) M. Wada, T. Murata, H. Oikawa and H. Oguri, Org. Biomol. Chem., 2014, 12, 298–306 RSC.
- (a) R. H. Snell, R. L. Woodward and M. C. Willis, Angew. Chem., Int. Ed., 2011, 50, 9116–9119 CrossRef CAS PubMed; (b) H. Ishikawa, H. Takayama and N. Aimi, Tetrahedron Lett., 2002, 43, 5637–5639 CrossRef CAS; (c) L. Verotta, F. Orsini, M. Sbacchi, M. A. Scheildler, T. A. Anador and E. Elisabetsky, Bioorg. Med. Chem., 2002, 10, 2133–2142 CrossRef CAS; (d) M. Nagakawa, H. Sugumi, S. Kodato and T. Hino, Tetrahedron Lett., 1981, 22, 5323–5326 CrossRef; (e) T. Hino, S. Kodato, K. Takahashi, H. Yamaguchi and M. Nakagawa, Tetrahedron Lett., 1978, 19, 4913–4916 CrossRef.
- (a) S. Tadano, Y. Mukaeda and H. Ishikawa, Angew. Chem., Int. Ed., 2013, 52, 7990–7994 CrossRef CAS PubMed; (b) Y.-X. Li, H.-X. Wang, S. Ali, X.-F. Xia and Y.-M. Liang, Chem. Commun., 2012, 48, 2343–2345 RSC; (c) E. S. Hall, F. McCapra and A. I. Scott, Tetrahedron, 1967, 23, 4131–4141 CrossRef CAS; (d) A. I. Scott, F. McCapra and E. S. Hall, J. Am. Chem. Soc., 1964, 86, 302–303 CrossRef CAS.
- (a) T. Araki, Y. Manabe, K. Fujika, H. Yokoe, M. Kanematsu, M. Yoshida and K. Shishido, Tetrahedron Lett., 2013, 54, 1012–1014 CrossRef CAS PubMed; (b) R. Liu and J. Zhang, Org. Lett., 2013, 15, 2266–2269 CrossRef CAS PubMed; (c) W. Xie, H. Wang, F. Fan, J. Tian, Z. Zuo, W. Zi, K. Gao and D. Ma, Tetrahedron Lett., 2013, 54, 4392–4396 CrossRef CAS PubMed; (d) M. Movassaghi, O. K. Ahmad and S. P. Lathrop, J. Am. Chem. Soc., 2011, 133, 13002–13005 CrossRef CAS PubMed; (e) S. P. Lathrop and M. Movassaghi, Chem. Sci., 2014, 5, 333–340 RSC.
- R. Robinson and H. J. Teuber, Chem. Ind., 1954, 783–784 CAS.
- (a) M. Ohno, T. F. Spande and B. Witkop, J. Am. Chem. Soc., 1970, 92, 343 CrossRef CAS; (b) A. S. Cardoso, N. Srinivasan, A. M. Lobo and S. Prabhakar, Tetrahedron Lett., 2001, 42, 6663 CrossRef CAS; (c) M. Okada, I. Sato, S. J. Cho, D. Dubnau and Y. Sakagami, Tetrahedron, 2006, 62, 8907 CrossRef CAS PubMed.
- For examples on indole-involved, I2-mediated oxidative coupling reactions, see: (a) Z. Zuo, W. Xie and D. Ma, J. Am. Chem. Soc., 2010, 132, 13226 CrossRef CAS PubMed; (b) Z. Zuo and D. Ma, Angew. Chem., Int. Ed., 2011, 50, 12008 CrossRef CAS PubMed; (c) W. Zi, W. Xie and D. Ma, J. Am. Chem. Soc., 2012, 134, 9126 CrossRef CAS PubMed.
- CCDC references numbers 1012369 (6) and 1012368 (7).
- CCDC reference number 1012367.
- The reaction of 15 with LiHMDS/I2 in THF at −40 °C also afforded 16, but in a low yield (∼40%). Apparently the effect of the C2-ester moiety on 15 prevailed in controlling the stereoselectivity of dimerization.
- L. A. Carpino, J. Am. Chem. Soc., 1993, 115, 4397–4398 CrossRef CAS.
- Lera et al. synthesized 3 from 17 in 3 steps and the last step had to be performed at 180 °C (ref. 7a). Ishikawa et al. reported the one-step synthesis of 3 from the ethyl ester analog of 17 (ref. 11a). However, it was run at 230 °C under vacuum. Compared to their methods, our conditions are much milder (RT for both steps).
- For selected examples on the decarboxylation of tryptophan derivatives, see: (a) M. Bruncko, D. Crich and R. Samy, J. Org. Chem., 1994, 59, 5543–5549 CrossRef CAS; (b) P. Chen, L. Cao, W. Tian, X. Wang and C. Li, Chem. Commun., 2010, 46, 8436–8438 RSC; (c) D. Sun, Q. Zhao and C. Li, Org. Lett., 2011, 13, 5302–5305 CrossRef CAS PubMed.
- (a) A. D. Lebsack, J. T. Link, L. E. Overman and B. A. Stearms, J. Am. Chem. Soc., 2012, 124, 9008–9009 CrossRef PubMed; (b) L. E. Overman and E. A. Peterson, Angew. Chem., Int. Ed., 2003, 42, 2525–2528 CrossRef CAS PubMed; (c) J. Kodanko and L. E. Overman, Angew. Chem., Int. Ed., 2003, 42, 2528–2531 CrossRef CAS PubMed; (d) R. H. Snell, M. J. Durbin, R. L. Woodward and M. C. Willis, Chem. – Eur. J., 2012, 18, 16754–16764 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1012367–1012369. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00165f |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2014 |