A convenient, economical and scalable multi-gram synthesis of 1-vinylcyclopropyl 4-methylbenzenesulfonate†‡
O. Stephen
Ojo
a,
Phillip A.
Inglesby
a,
Daniela E.
Negru
b and
P. Andrew
Evans
*b
aDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK
bDepartment of Chemistry, Queen's University, 90 Bader Lane, Kingston, ON K7L 3N6, Canada. E-mail: Andrew.Evans@chem.queensu.ca
First published on 1st July 2014
Abstract
We describe a practical, economical, and scalable multi-gram synthesis of 1-vinylcyclopropyl 4-methylbenzenesulfonate. This intermediate is important for the rapid and economical synthesis of alkylidenecyclopropanes, which provides access to a variety of alicyclic systems via metal-catalysed higher-order carbocyclization and cycloisomerization reactions.
1-Vinylcyclopropyl 4-methylbenzenesulfonate (1) represents a particularly important synthetic intermediate for the construction of carbon- and heteroatom-tethered alkylidenecyclopropanes (ACPs) 2.1–3 ACPs provide versatile three-carbon synthons for a variety of metal-catalyzed cycloaddition reactions and as such are extremely important intermediates.3 For example, this unit has featured in the palladium-catalyzed [(3 + 2)] and [(4 + 3)] carbocyclization reactions to afford 3 and 4, respectively (Scheme 1).3,4 More recently, we reported the rhodium-catalyzed [(3 + 2) + 2] and [(3 + 2) + 1] carbocyclizations of 2 with alkynes and carbon monoxide to afford 5 and 6.5,6 Furthermore, the ACP 2 (R1 = CH2OH) undergoes a facile ene-cycloisomerization reaction (ECI) to generate the five-membered ring 7, thereby illustrating significant versatility of this motif.7,8
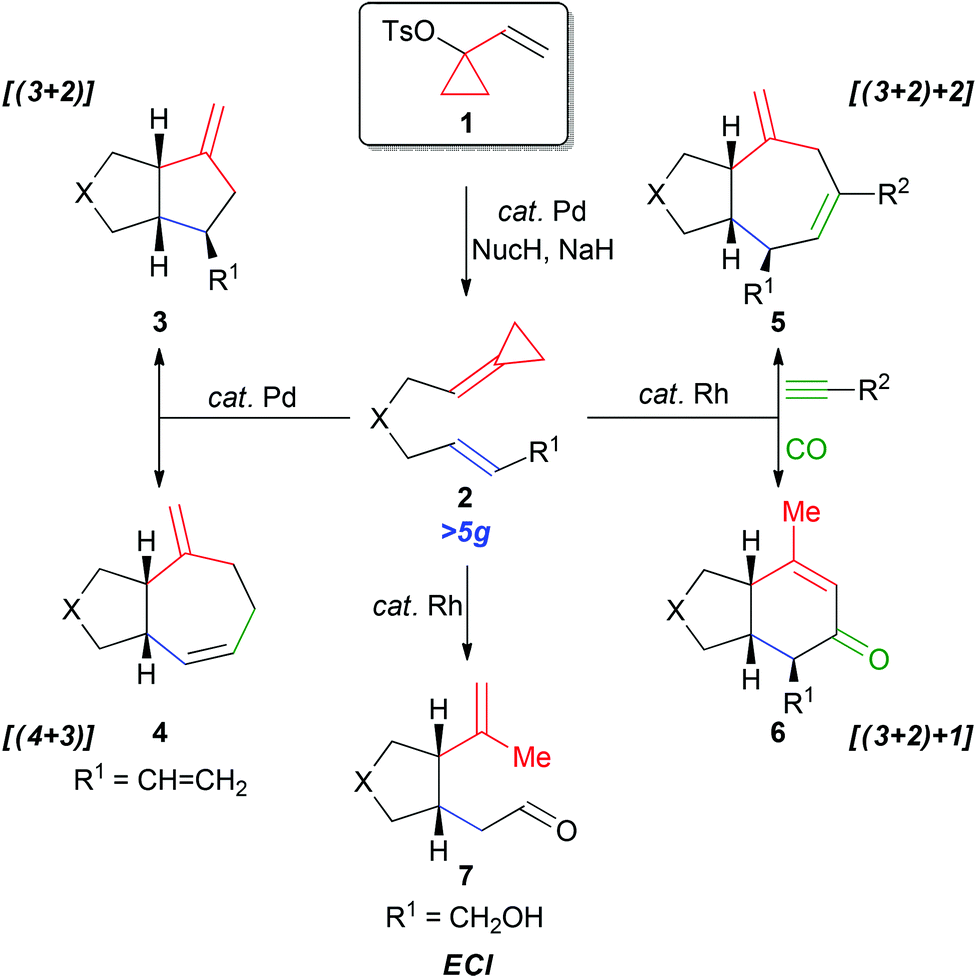 | ||
| Scheme 1 Synthetic utility of VCPs and ACPs (X = O, NTs, C(CO2Me)2). | ||
Since many ACPs are prepared directly from 1-vinylcyclopropyl 4-methylbenzenesulfonate (1) via a palladium-catalyzed Tsuji–Trost allylic alkylation with carbon and heteroatom nucleophiles, the ability to access suitable quantities of 1 in an affordable and efficient manner is important.9,10 Preliminary studies centred on the synthesis of a variety of derivatives of ACP 2 from VCP 1, highlighted a number of significant limitations with the preparation of this important intermediate (Scheme 2A). For example, VCP 1 can be prepared via protodesilylation of 8 followed by Grignard addition to the ketone and tosylation of the tertiary alcohol (Method A).11 Alternatively, the oxidation and tosyl protection of 9, followed by silyl deprotection and partial alkyne reduction provides the ACP 1 (Method B).12 Nevertheless, the ability to prepare decagram quantities of 1 was hampered by, (1) the high cost of the precursors 8 and 9,13–15 (2) the difficult separation of 1 from unreacted p-toluenesulfonyl chloride (Method A), and (3) low temperature conditions (Method B), which in turn impacted our ability to develop new and exciting reactions involving ACPs. Despite the growing popularity of ACPs over the last decade, the limitations in the preparation of VCP 1 provides the impetus for the development of a practical and economical synthesis of this intermediate. Herein, we now report a scalable synthesis of VCP 1 from a cheap and readily available precursor, namely ethyl 3-chloropropionate 10, using a 3-step synthetic sequence outlined in Scheme 2B.
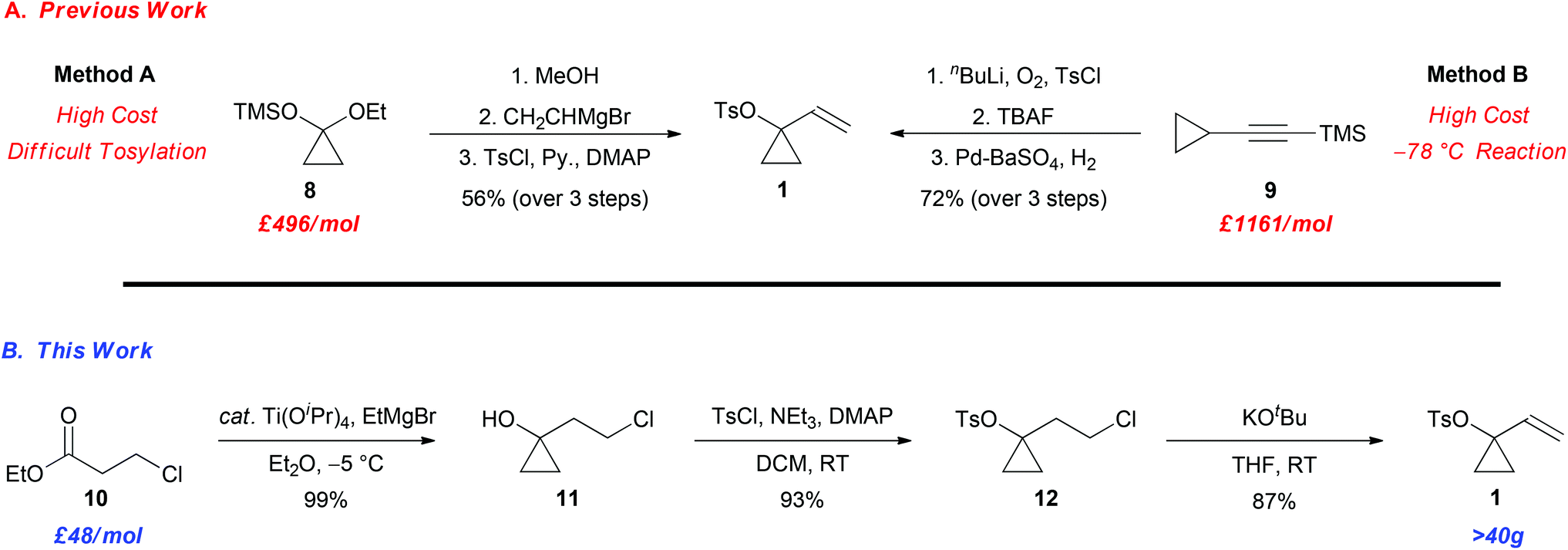 | ||
| Scheme 2 A comparison of the methods utilized for the preparation of 1-vinylcyclopropyl-4-methylbenzenesulfonate (1). | ||
The synthesis of 1 commenced with the known Kulinkovich cyclopropanation of the ethyl ester 10 using catalytic titanium(IV) isopropoxide and ethylmagnesium bromide to afford the cyclopropanol 11 in 99% yield (Scheme 2B).16,17 Tosylation of the tertiary cyclopropyl alcohol using p-toluenesulfonyl chloride, triethylamine and catalytic 4-dimethylaminopyridine (DMAP) furnished intermediate 12 in 93% yield. The tosylation of the tertiary alcohol proceeds smoothly in this case, which contrasts the aforementioned strategy wherein the tosylation of the tertiary allylic alcohol is more problematic (Scheme 1A; Method A). The sequence was then completed with an E2 elimination of the primary alkyl chloride with potassium tert-butoxide to furnish the desired VCP 1 in 87% yield (80% overall yield for 3 steps).18,19 Gratifyingly, the synthesis can be performed on 40 g scale and all reactions are performed between −5 °C and room temperature. Hence, this operationally simple protocol provides a convenient and scalable sequence for the construction of VCP 1, which is likely to prove useful to others to facilitate additional studies in this exciting area of research.
Procedures20
1-(2-Chloroethyl)cyclopropanol (11)
To a flame-dried 2 L three-necked round-bottomed flask, equipped with a magnetic stirrer bar, thermometer and a pressure equalizing dropping funnel was added ethyl 3-chloropropanoate 10 (30.0 g, 220 mmol), anhydrous diethyl ether (700 mL) and titanium isopropoxide (13.01 mL, 43.9 mmol). The system was purged with a continuous flow of argon and the reaction mixture was cooled with stirring to −5 °C using a salt-ice bath. The addition funnel was charged with ethylmagnesium bromide (146 mL, 439 mmol; 3 M in diethyl ether), which was added dropwise to the stirred reaction mixture at a rate to maintain the internal reaction temperature below +5 °C (ca. 30 minutes). After the addition was complete, the salt-ice bath was removed and the reaction mixture was warmed to room temperature and stirred for ca. 2 hours (t.l.c. control). The reaction was quenched by addition of a 1 M aqueous hydrochloric chloride solution (300 mL) over 20 minutes using an ice bath to maintain the reaction temperature between 20–25 °C. The mixture was then poured into a 3 L separatory funnel, diluted with deionized water (500 mL) and partitioned with diethyl ether (2 × 500 mL). The organic phases were combined, washed with saturated aqueous sodium bicarbonate solution (500 mL), saturated aqueous NaCl solution (200 mL), dried (anhyd. MgSO4), filtered and concentrated in vacuo to provide 1-(2-chloroethyl)cyclopropanol 11 as a colorless oil (26.45 g, 219 mmol, 99%), which was used without further purification.1-(2-Chloroethyl)cyclopropyl 4-methylbenzenesulfonate (12)
To a flame-dried 2 L two-necked round-bottomed flask, equipped with a magnetic stirrer bar, was added 1-(2-chloroethyl)cyclopropanol 11 (26.45 g, 219 mmol) and anhydrous dichloromethane (700 mL) under an atmosphere of argon. Triethylamine (36.6 mL, 263 mmol) was slowly added to the stirred reaction mixture followed by 4-dimethylaminopyridine (26.7 g, 219 mmol) in one portion. The reaction mixture was stirred at room temperature for ca. 30 minutes before p-toluenesulfonyl chloride (55.4 g, 285 mmol) was added and the resulting mixture stirred for ca. 16 hours at room temperature. The reaction mixture was concentrated in vacuo to a volume of approximately 100 mL, quenched with deionized water (500 mL) and transferred to a 3 L separatory funnel. The aqueous phase was partitioned with diethyl ether (500 mL), diluted with saturated aqueous ammonium chloride (350 mL) and further partitioned with diethyl ether (2 × 350 mL). The combined organic phases were washed with deionized water (250 mL), dried (anhyd. MgSO4), filtered and concentrated in vacuo (35 °C, 1 torr) to afford the crude product. The crude material was dry-packed onto silica gel and purified via flash chromatography (SiO2, eluting with 5–15% diethyl ether–petroleum ethers) to afford 1-(2-chloroethyl)cyclopropyl 4-methylbenzenesulfonate 12 (56.50 g, 206 mmol, 93%) as a pale green oil.1-Vinylcyclopropyl 4-methylbenzenesulfonate (1)
To a flame-dried 2 L one-necked round-bottom flask, equipped with a magnetic stirrer bar was added 1-(2-chloroethyl)cyclopropyl 4-methylbenzenesulfonate 12 (56.5 g, 206 mmol) and anhydrous tetrahydrofuran (700 mL) under an atmosphere of argon. Potassium tert-butoxide (35.7 g, 308 mmol) was added at room temperature in one portion to the stirred solution, and the resulting suspension was stirred for an additional ca. 2 hours (monitored by NMR). The reaction mixture was concentrated in vacuo to a volume of approximately 500 mL, diluted with deionized water (300 mL), transferred into a 2 L separatory funnel and partitioned with diethyl ether (350 mL). The aqueous layer was diluted with saturated aqueous ammonium chloride solution (200 mL) and further partitioned with diethyl ether (2 × 300 mL). The combined organic phases were washed with deionized water (300 mL), saturated aqueous NaCl solution (200 mL), dried (anhyd. MgSO4), filtered and concentrated in vacuo to afford the crude product. The crude material was dry-packed onto silica gel and purified via flash chromatography (SiO2, eluting with 5–10% diethyl ether–petroleum ethers) to afford 1 (42.40 g, 178 mmol, 87%) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.78 (d, J = 8.3 Hz, 2H), 7.32 (d, J = 8.2 Hz, 2H), 5.89 (dd, J = 17.2, 10.8 Hz, 1H), 5.10 (d, J = 17.2 Hz, 1H), 5.01 (d, J = 11.1 Hz, 1H), 2.45 (s, 3H), 1.36 (d, A of AB, JAB = 6.3 Hz, 1H), 1.35 (d, B of AB, JAB = 6.1 Hz, 1H), 0.92 (dd, A of ABM, JAB = 5.6 Hz, JAM = 1.0 Hz, 1H), 0.91 (dd, B of ABM, JAB = 6.0 Hz, JBM = 0.8 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 144.84 (e), 136.62 (o), 135.01 (e), 129.83 (o), 127.94 (o), 113.52 (e), 65.47 (e), 21.73 (o), 14.04 (e); IR (Neat) 3003 (w), 1644 (w), 1596 (w), 1349 (vs), 1294 (m), 1165 (vs), 1089 (s), 1032 (s), 949 (vs), 906 (vs), 819 (vs) cm−1; HRMS (ESI, [M + H]+) calculated for C12H15O3S 239.0736, found 239.0729.References
- (a) H. H. Wasserman, R. E. Cochoy and M. S. Baird, J. Am. Chem. Soc., 1969, 91, 2375 CrossRef CAS; (b) B. A. Howell and J. G. Jewett, J. Am. Chem. Soc., 1971, 93, 798 CrossRef.
- For a recent review on thermal- and metal-promoted rearrangements of VCPs and their derivatives, see: T. Hudlicky and J. W. Reed, Angew. Chem., Int. Ed., 2010, 49, 4864 Search PubMed.
- For reviews on ACPs, see: (a) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed; (b) A. Masarwa and I. Marek, Chem. – Eur. J., 2010, 16, 9712 CrossRef CAS PubMed; (c) H. Pellissier, Tetrahedron, 2010, 66, 8341 CrossRef CAS PubMed; (d) C. Aïssa, Synthesis, 2011, 3389 CrossRef PubMed.
- For selected examples of palladium-catalyzed [(3 + 2)] and [(4 + 3)] carbocyclizations of ACPs, see: (a) S. Yamago and E. Nakamura, J. Chem. Soc., Chem. Commun., 1988, 1112 RSC; (b) M. Gulías, R. García, A. Delgado, L. Castedo and J. L. Mascareñas, J. Am. Chem. Soc., 2006, 128, 384 CrossRef PubMed; (c) M. Gulías, J. Durán, F. López, L. Castedo and J. L. Mascareñas, J. Am. Chem. Soc., 2007, 129, 11026 CrossRef PubMed and pertinent references cited therein.
- For selected reviews on transition metal-catalyzed [m + n + o] carbocyclization reactions, see: (a) P. A. Inglesby and P. A. Evans, Chem. Soc. Rev., 2010, 39, 2791 RSC; (b) C. Aubert, M. Malacria and C. Ollivier, in Science of Synthesis, ed. J. G. De Vries, G. A. Molander and P. A. Evans, Thieme, Stuttgart, 2011, vol. 3, pp. 145–242 and pertinent references cited therein Search PubMed.
- (a) P. A. Evans and P. A. Inglesby, J. Am. Chem. Soc., 2008, 130, 12838 CrossRef CAS PubMed; (b) S. Mazumder, D. Shang, D. E. Negru, M.-H. Baik and P. A. Evans, J. Am. Chem. Soc., 2012, 134, 20569 CrossRef CAS PubMed; (c) P. A. Inglesby, J. Basca, D. E. Negru and P. A. Evans, Angew. Chem., Int. Ed., 2014, 53, 3952 CrossRef CAS PubMed.
- For a recent review on transition metal-catalyzed ene-cycloisomerization reactions, see: I. D. G. Watson and F. D. Toste, in Science of Synthesis, ed J. G. De Vries, G. A. Molander and P. A. Evans, Thieme, Stuttgart, Germany, 2011, vol. 3, pp. 243–307 and pertinent references cited therein Search PubMed.
- P. A. Evans and P. A. Inglesby, J. Am. Chem. Soc., 2012, 134, 3635 CrossRef CAS PubMed.
- For seminal work on the preparation of ACPs using palladium-catalyzed Tsuji–Trost alkylations, see: (a) A. Stolle, J. Salaün and A. de Meijere, Tetrahedron Lett., 1990, 31, 4593 CrossRef CAS; (b) A. Stolle, J. Salaün and A. de Meijere, Synlett, 1991, 327 CrossRef CAS PubMed; (c) A. Stolle, J. Ollivier, P. P. Piras, J. Salaün and A. de Meijere, J. Am. Chem. Soc., 1992, 114, 4051 CrossRef CAS and pertinent references cited therein.
- For an example of umpolung additions of the π-allyl intermediate derived from 1, see: J. Ollivier, N. Girard and J. Salaün, Synlett, 1999, 1539 CrossRef CAS PubMed.
- For the synthesis of 1 using Method A, see ref. 9c and pertinent references cited therein.
- For the synthesis of 1 using Method B, see: S. Bräse, S. Schömenauer, G. McGaffin, A. Stolle and A. de Meijere, Chem. – Eur J., 1996, 2, 545 CrossRef.
- Pricing of chemicals were obtained on 28th February 2014 and are as follows: 8 from Acros, 9 from Fluorochem (Oakwood) and 10 from Alfa Aesar. The prices were selected by virtue of the lowest £ mol−1 and does not account for the quantity purchased or reagent purity.
- Although (1-ethoxycyclopropoxy)trimethylsilane 8 can be prepared from 10; this approach adds an additional step and does not circumvent the aforementioned challenges.15.
- P. A. Wender, A. J. Dyckman, C. O. Husfeld and M. J. C. Scanio, Org. Lett., 2000, 2, 1609 CrossRef CAS PubMed.
- O. G. Kulinkovich, Chem. Rev., 2003, 103, 2597 CrossRef CAS PubMed.
- O. G. Kulinkovich, Y. Y. Kozyrkov, A. V. Bekish, E. A. Matiushenkov and I. L. Lysenko, Synthesis, 2005, 1713 CrossRef CAS PubMed.
- It is noteworthy that these conditions have been previously employed to prepare substituted VCPs, however they have not been used to specifically prepare VCP (1): (a) I. Sylvestre, J. Ollivier and J. Salaün, Tetrahedron Lett., 2001, 42, 4991 CrossRef CAS; (b) S. Racouchot, I. Sylvestre, J. Ollivier, Y. Y. Kozyrkov, A. Pukin, O. G. Kulinkovich and J. Salaün, Eur. J. Org. Chem., 2002, 2160 CrossRef CAS.
- Similar yields were obtained in 3 separate experiments on this scale.
- See ESI‡ for full details of experimental procedures, structural analyses and reagent specifications.
Footnotes |
| † Dedicated to Professor Max Malacria on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Representative procedures, spectral data and copies of spectra. See DOI: 10.1039/c4qo00089g |
| This journal is © the Partner Organisations 2014 |