 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-sorting of crown ether/secondary ammonium ion hetero-[c2]daisy chain pseudorotaxanes†
Bo
Zheng
a,
Fabian
Klautzsch
b,
Min
Xue
a,
Feihe
Huang
a and
Christoph A.
Schalley
*b
aState Key Laboratory of Chemical Engineering, Department of Chemistry, Zhejiang University, Hangzhou, Zhejiang 310027, China. E-mail: fhuang@zju.edu.cn
bInstitut für Chemie und Biochemie, Freie Universität Berlin, Takustr. 3, 14195, Berlin, Germany. E-mail: c.schalley@fu-berlin.de
First published on 21st April 2014
Abstract
Four monomeric building blocks equipped with one crown ether and one secondary ammonium ion are synthesized and studied with respect to their ability to form daisy chain dimers. Two crown ethers with different cavity sizes – i.e. [21]crown-7 and [24]crown-8 – and two ammonium ions substituted with either a thin alkyl group or a more bulky benzyl group are used as the binding motifs. Self-sorting behaviour can be expected as the [21]crown-7/alkyl ammonium and [24]crown-8/benzyl ammonium binding motifs are orthogonal. Three homodimers are characterized by NMR, X-ray crystallography and ESI mass spectrometry. They are recognizable by the presence of signals for diastereotopic protons in the 1H NMR spectra. The formation of hetero-[c2]daisy chain dimers can be monitored by NMR spectroscopy and ESI mass spectrometry and show the expected self-sorting behaviour.
Introduction
Nature efficiently and successfully assembles intricate and highly complex architectures,1 in which high-fidelity self-sorting2 is ubiquitous as the basis of the building blocks’ cooperative functional interplay. Many supramolecular chemists have recently constructed synthetic self-sorting systems with orthogonal binding motifs fabricated on the basis of hydrogen bonding,3 metal–ligand interactions,4 π–π-stacking5 or solvophobic effects.6 A number of different factors, among them the orthogonality of the recognition motifs, size, shape, thermodynamic and kinetic binding parameters, stoichiometry and concentration define the molecular codes and are responsible for self-recognition (narcissistic self-sorting) or self-discrimination (social self-sorting).2,5a,7Recently, we have applied the concepts of social self-sorting to pseudorotaxane8 assemblies and supramolecular pseudorotaxane polymers based on the crown ether/secondary ammonium ion binding motifs.9 Benzo[21]crown-7 C7 and dibenzo[24]crown-8 C8 (Fig. 1) have two different cavity sizes and their pseudorotaxane binding motifs with secondary ammonium ions can be made orthogonal by using either benzyl-n-butyl ammonium BBA with one thin alkyl ammonium or dibenzyl ammonium DBA with two more bulky benzyl ammonium stopper groups. As the benzyl stopper is too large to penetrate C7, only three host–guest complexes BBA·C7, BBA·C8 and DBA·C8 can form. Fig. 1 summarizes the binding constants for these pairs in a CDCl3–CH3CN (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solvent mixture. When the feed ratio is controlled and all four components are mixed in equimolecular amounts, self-sorting is obtained, because the DBA guest cation consumes all C8 host molecules while only C7 binds BBA. But even, when feed ratio is not perfectly matched, this self-sorting behaviour is expected to occur, because the BBA·C7 and DBA·C8 pairs exhibit the two highest binding constants of 14500 M−1 and 5500 M−1, respectively, among the three possible pseudorotaxanes.10 Consequently, the resulting self-sorting is driven kinetically (stopper sizes) as well as thermodynamically (binding constants) into the same direction.
:1) solvent mixture. When the feed ratio is controlled and all four components are mixed in equimolecular amounts, self-sorting is obtained, because the DBA guest cation consumes all C8 host molecules while only C7 binds BBA. But even, when feed ratio is not perfectly matched, this self-sorting behaviour is expected to occur, because the BBA·C7 and DBA·C8 pairs exhibit the two highest binding constants of 14500 M−1 and 5500 M−1, respectively, among the three possible pseudorotaxanes.10 Consequently, the resulting self-sorting is driven kinetically (stopper sizes) as well as thermodynamically (binding constants) into the same direction.
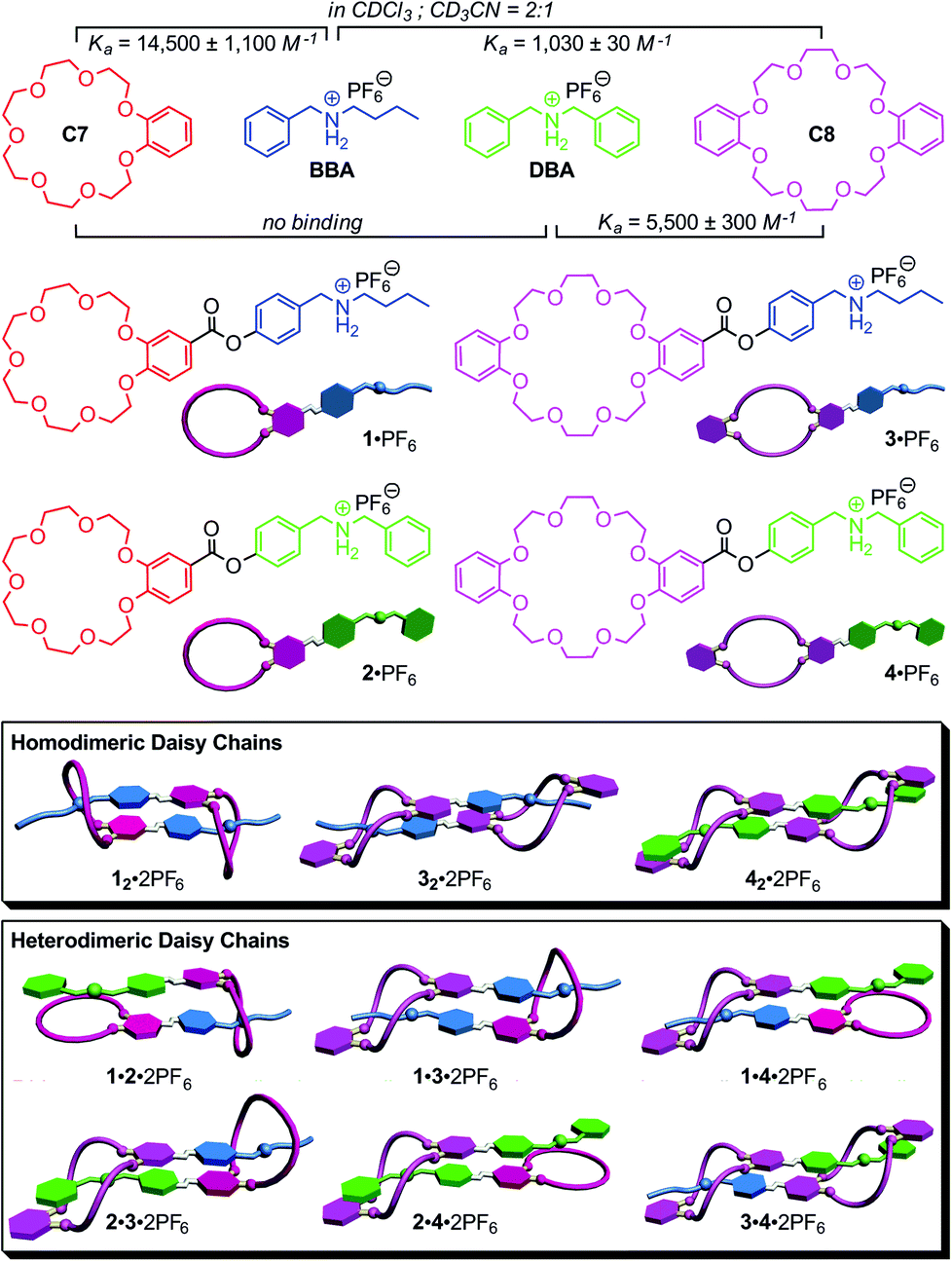 | ||
| Fig. 1 Top: Structures and binding data of building blocks C7, C8, BBA and DBA. Centre: Monomers 1·PF6–4·PF6. Bottom: Cartoons representing all possible homo- and heterodimeric daisy chains. Note that dimer 22·2PF6 does not form and 1·2·2PF6 and 1·4·2PF6 cannot ring-close. | ||
[c2]Daisy chains are cyclic interwoven oligo- or even polymers11 that are usually constructed from AB-type plerotopic monomers equipped with two complementary binding units A (host) and B (guest). Usually, small, often dimeric macrocyclic assemblies result, because they are enthalpically (all binding sites involved in binding) as well as entropically (high particle number) favourable. Such self-complementary building blocks – when equipped with two switchable stations – are also excellent building blocks for molecular muscle-type supramolecular polymers.12 The daisy chains known so far are all constructed from identical monomers. This limits the structural diversity that can be obtained by self-assembly. In contrast, progress towards functional complexes containing different cooperating building blocks to realize high-level function would require the ability to build well-defined architectures, in which each subunit occupies a precisely controlled position. It is therefore highly desirable to provide strategies for programming different monomers into heteromeric assemblies. Self-sorting is one potential strategy in this direction.
Here, we apply the principles of social self-sorting to the construction of heterodimeric [c2]daisy chains. The aim of the present study is to investigate their self-sorting behaviour and to extend the scope of the self-assembly of hierarchical heteromeric assemblies. The investigation of heteromeric assemblies helps understand self-sorting better and will contribute to paving a way to build well-defined assemblies that carry cooperating groups for implementing function. Four different, yet structurally quite similar monomers 1·PF6–4·PF6 were designed (Fig. 1, centre) based on the two different crown ethers C7 and C8 and two different secondary ammonium ions BBA and DBA. In order to obtain these four AB-type monomers, the two binding sites – one crown ether and one ammonium ion – are connected through ester linkages as reported earlier for 1·PF6.13
Based on the self-sorting behaviour of C7, C8, BBA and DBA, one would arrive at the following expectations (Fig. 1, bottom). As 2·PF6 is not self-complementary due to the combination of the larger benzyl stopper with smaller crown, this monomer is predicted not to dimerize. In solutions of the pure monomers, three homodimers should therefore form under the appropriate conditions: 12·2PF6, 32·2PF6 and 42·2PF6. A 1:1 mixture of 2·PF6 and 3·PF6 should convert fully into the heterodimer 2·3·2PF6 (perfect self-sorting), because the two building blocks are matching each other. Binding can only involve all binding sites, when the heterodimeric daisy chain forms exclusively. Otherwise, monomeric 2·PF6 would remain with unsaturated binding sites along with the 32·2PF6 homodimer. For a 1:1 mixture of 1·PF6 and 3·PF6, one predicts a statistical 1:2:1 mixture of 12·2PF6, 1·3·2PF6 and 32·2PF6 as the final result of the assembly, since both homodimers as well as the heterodimer can easily form and their equilibration should be more or less thermoneutral. For all other combinations, no significant heterodimer formation is expected, as the heterodimers 1·2·2PF6, 1·4·2PF6 and 2·4·2PF6 can merely form singly threaded heterodimeric daisy chains. The formation of heterodimers would thus lead to unsaturated binding sites and is then less favourable as the mixture of both homodimers.
Results and discussion
Monomer synthesis and characterisation
The synthesis of 2·PF6–4·PF6 follows the previously reported preparation of 1·PF6 (Fig. 2).13,14 Briefly, the acid derivatives of the two crown ethers, C7-COOH and C8-COOH, were coupled with Boc-protected derivatives 5 and 6 of DBA and BBA, respectively. After de-protection and ion exchange, the desired target compounds 1·PF6–4·PF6 were obtained.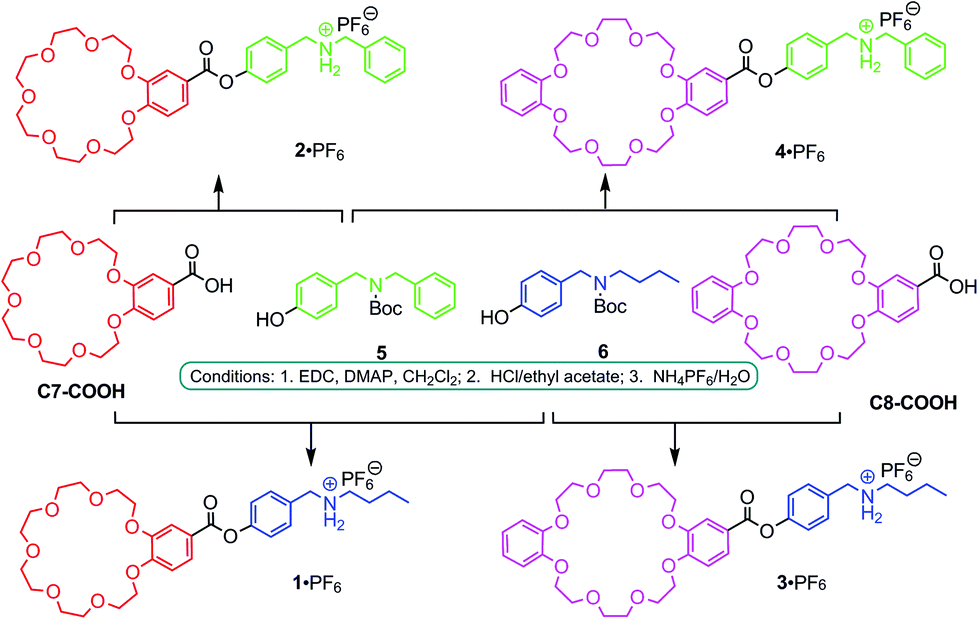 | ||
| Fig. 2 Synthesis of monomers 1·PF6–4·PF6. (EDC = 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride; DMAP = 4-dimethylaminopyridine). | ||
In order to characterize the samples as monomers, NMR spectra were recorded in DMSO-d6 (ESI†), in which only one set of signals is observed. Although the substitution of the crown ethers’ benzo moieties with the ester group renders all methylene groups of the crowns non-equivalent, they are somewhat broadened as they are pairwise almost isochronous and thus closely overlapping mostly depending on the distance to the aromatic ring.
Homodimers 12·2PF6, 32·2PF6 and 42·2PF6
Before discussing the formation of homodimers, a brief comment on the chirality features of the daisy chains may be indicated. All four monomers are achiral compounds. Upon threading an unsymmetrical axle through each of the crown ethers, chiral complexes are possible. The ester groups define directionality around the crown ether; the axle defines a direction perpendicular to the crown ether plane. Consequently, such a complex exhibits planar chirality. This is certainly true for all heterodimeric [c2]daisy chains, which may thus form four different stereoisomers that are in equilibrium with each other. However, one should be aware of the fact that homodimeric [c2]daisy chains may exist as a pair of enantiomers plus one achiral meso-diastereomer bearing a centre of inversion so that only three stereoisomers can exist in equilibrium here.In less competitive solvents, three of the monomers, 1·PF6,133·PF6 and 4·PF6, are expected to form homodimers. Indeed, the 1H NMR spectra of these three compounds in a 2:1 mixture of chloroform and acetonitrile (Fig. 3) are much more complex than those in DMSO, while 2·PF6 retains its simplicity also in this less competitive solvent mixture. The increased complexity is caused by two effects: threading on one hand leads to more significant differences in the environments of those crown ether methylene groups that overlap in the spectra of the monomers. In addition, the two faces of the crown ethers become different upon threading of the axle. Thus, at least some of the methylene groups give rise to separate signals for the two protons. Even with the help of 2D spectra, a complete assignment of all signals is difficult as many of them still strongly overlap (ESI†). The best signal separation is found for 42·2PF6 (Fig. 3) so that we can clearly identify two pairs of diastereomers (one from the phenylmethyl proton and one from the crown methylene) and other diastereotopic signals of crown CH2 groups here (ESI†). Also, typical complexation-induced shifts indicate the formation of daisy chain dimers: In particular, the aromatic protons Ha and Hc of 12·PF6, 32·PF6 and 42·PF6 are shifted up-field relative to those of the free monomer in DMSO-d6, the monomer form of 2·PF6 in CDCl3–CD3CN (2:1) or the potassium complex (vide infra), with the most significant shift observed for Ha. This indicates – irrespective of the reference system chosen – the up-field shift to be due to the anisotropy effect of stacked aromatic rings which are only present in the daisy chain dimers, but do not exist in the monomers. Together with the observation of intense signals for the homodimers in the ESI mass spectra, the drastic changes in the 1H NMR spectra for 1·PF6, 3·PF6 and 4·PF6 and the absence of similar changes for 2·PF6 clearly indicate the formation of the expected homodimers. Even though thermodynamically controlled macrocyclisations can give rise to larger cyclic oligomers,15 no larger complexes than dimers were detected in the mass spectra so that we rule out significant contributions from larger oligomers to the equilibrium situation.
 | ||
| Fig. 3 Partial 1H NMR spectra (500 MHz, CDCl3–CD3CN (2:1) 10.0 mM, 298 K) of (a) 12·2PF6, (b) 2·PF6, (c) 32·2PF6 and (d) 42·2PF6. | ||
Another piece of evidence for the formation of homodimers is their switchability with a competitive guest. When KPF6 is added to the NMR solutions of the three homodimers (ESI†), the complex NMR spectra return into much simpler ones that again show only one set of signals. These signals of the potassium complex are shifted only very slightly relative to the uncomplexed monomer as demonstrated for example for 2·PF6 (ESI†).
Although all attempts to crystallize the heterodimers under study failed unfortunately, single crystals of homodimers 32·2PF6 and 42·2PF6 were obtained. These solid state structures are shown in Fig. 4a, c and e together with the previously reported crystal structure of 12·2PF6,13 which is added to the figure to facilitate direct comparison. It is clear that in the crystal structures all three homodimers are doubly-threaded head-to-tail daisy chains. The ammonium ions are positioned inside the cavities of the crown ethers by multiple N–H⋯O and C–H⋯O hydrogen bonds. Table 1 summarizes the hydrogen bonding distances and angles for N–H⋯O and C–H⋯O hydrogen bonds which are in the ranges expected for crown ether/ammonium binding motifs. Apparently, the formation of dimers is also supported by face-to-face π-stacking interaction between the aromatic ring of the host moiety and the phenyl ring of the ammonium salt unit. The centroid–centroid distances are 3.64 Å in 12·2PF6 and 3.68 Å in 32·2PF6. The structure of 42·2PF6 differs in three respects from the other two: (i) although both monomers in the homodimer are identical, its crystal structure reveals two different binding motifs with different hydrogen bonding parameters realized – an effect that is likely due to packing effects. (ii) The aromatic rings are not aligned in a face-to-face fashion, but are twisted into arrangements with inclinations of 70.1° and 82.2° between the two planes of adjacent aromatic rings. (iii) Both ester carbonyl groups point in the same direction, while the other two homodimers have ester groups that point in opposite directions. Consequently, 12·2PF6 and 32·2PF6 bear a centre of inversion and realize the above-mentioned meso-forms, while no centre of inversion exists in the crystal of 42·2PF6, which thus is present as a chiral complex. The daisy chain homodimers pack in the crystal to yield infinite supramolecular tapes driven by face-to-face π-stacking interactions (Fig. 4b, d and f). In the crystal structure of 42·2PF6, homochiral tapes each containing only one of the two enantiomeric assemblies alternate.
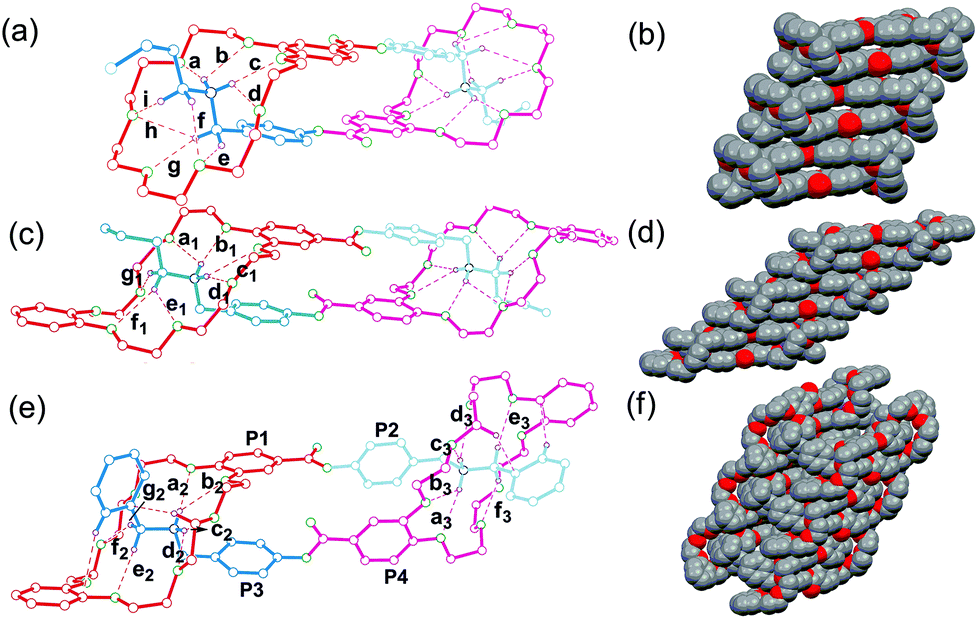 | ||
| Fig. 4 Left: Ball-stick views of the X-ray structures of (a) 12·2PF6, (c) 32·2PF6 and (e) 42·2PF6. PF6− counterions and hydrogen atoms except the ones involved in hydrogen bonding are omitted for clarity. Right: Space-filling representations of the packing of (b) 12·2PF6, (d) 32·2PF6 and (f) 42·2PF6. | ||
| 1 2·2PF6 (a–i) | 3 2·2PF6 (a1–g1) | 4 2·2PF6 (a2–g2) | 4 2·2PF6 (a3–f3) | ||
|---|---|---|---|---|---|
| a | d H⋯O | 2.46(3) | 2.49(5) | 2.32(4) | 2.40(8) |
| d N⋯O | 3.12(5) | 3.20(2) | 3.14(5) | 3.32(1) | |
| Angle | 130.6(4) | 129.4(6) | 139.7(6) | 170.7(2) | |
| b | d H⋯O | 2.16(6) | 2.29(0) | 2.56(5) | 2.58(7) |
| d N⋯O | 2.98(3) | 3.14(0) | 3.47(1) | 3.11(1) | |
| Angle | 150.5(7) | 153.3(8) | 168.4(4) | 116.6(7) | |
| c | d H⋯O | 2.25(3) | 2.40(2) | 2.46(9) | 2.05(3) |
| d N⋯O | 2.91(2) | 3.04(0) | 3.13(1) | 2.96(2) | |
| Angle | 129.7(6) | 126.5(4) | 129.0(0) | 169.6(7) | |
| d | d H⋯O | 2.11(2) | 1.99(6) | 2.05(0) | 2.56(6) |
| d N⋯O | 2.91(6) | 2.86(5) | 2.91(7) | 3.11(8) | |
| Angle | 148.2(0) | 157.0(0) | 156.9(2) | 119.0(4) | |
| e | d H⋯O | 2.61(4) | 2.64(5) | 2.33(8) | 2.49(9) |
| d C⋯O | 3.39(7) | 3.45(6) | 2.97(5) | 3.48(1) | |
| Angle | 138.0(0) | 139.0(5) | 126.2(6) | 171.0(6) | |
| f | d H⋯O | 2.29(5) | 2.56(0) | 2.51(5) | 2.35(6) |
| d C⋯O | 3.18(8) | 3.47(1) | 3.48(7) | 3.13(7) | |
| Angle | 152.7(0) | 152.8(6) | 167.0(6) | 135.1(1) | |
| g | d H⋯O | 2.71(1) | 2.62(4) | 2.55(1) | |
| d C⋯O | 3.15(9) | 3.59(8) | 3.43(0) | ||
| Angle | 108.7(2) | 167.7(6) | 147.9(3) | ||
| h | d H⋯O | 2.64(2) | |||
| d C⋯O | 3.49(9) | ||||
| Angle | 147.4(9) | ||||
| i | d H⋯O | 2.40(5) | |||
| d C⋯O | 3.28(0) | ||||
| Angle | 149.7(6) |
Mass spectrometry as a method to detect self-sorting
As discussed above, already the NMR spectra of the homodimers are rather complicated and difficult to interpret. If one attempts to analyse the generation of heterodimers, even more complicated spectra are expected to be observed. A detailed understanding, in particular a quantification of the contributions of homo- and heterodimers in solution will therefore not be straightforward by NMR spectroscopy. Crystallography neither provides a picture of the solution situation, even if suitable crystals could be grown, as solution equilibria may be shifted significantly upon crystallization. Therefore, ESI mass spectrometry appears to be a favourable tool to at least semi-quantitatively analyse the self-sorting behaviour of the daisy chains under study. A number of earlier studies on the assembly of other self-sorting pseudorotaxanes support this.3d,e,6b,9b,c,e,fFour points, however, may limit the scope of the analysis and should be considered in the interpretation of the data: (i) doubly charged homodimers will appear at the same m/z as singly charged monomers. They can however be easily distinguished as the peak spacing in the isotope patterns is 0.5 for the dicationic dimer and 1 for the singly charged monomer. Isotope pattern deconvolution has therefore been performed for all samples under study here. (ii) The sample solution concentrations used in ESI mass spectrometry are considerably lower than NMR concentrations. This may lead to a shift of the monomer–dimer equilibria towards the monomers so that the monomers may appear more prominently than expected from solution experiments. Furthermore, the monomers may form from the dimers, if the ions dissociate in part during ionization. Consequently, some contribution from monomeric species is expected to be observed. In the experiments reported here, sample concentrations were 100 μM in each monomer to reduce the concentration difference of NMR and MS samples as much as possible. (iii) Ion intensities do not necessarily directly reflect solution concentrations. The efficiencies with which the ions form during the electrospray process depend significantly on the desolvation energies16 which may vary for different structures. Although the differences in the so-called ESI response factors are often not very large for structurally similar ions, exceptions exist. (iv) Non-specific binding often occurs in ESI mass spectrometry. If it is prominent, it may blur the analysis of self-sorting, because non-specific complexes will not exhibit self-sorting and superimpose the true distribution of self-sorted complexes. In our study, we chose ionization conditions such that non-specific binding is minimal.
The 12·2PF6/42·2PF6 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1) and 2·PF6/42·2PF6 (2:1) mixtures
:1) and 2·PF6/42·2PF6 (2:1) mixtures
When 12·2PF6 and 42·2PF6 are mixed in equimolar amounts, no significant change is expected to occur. Both compounds form homodimers already in the stock solutions before mixing so that all binding sites are saturated. The heterodimer instead must remain singly threaded if it forms at all as the benzyl stopper in 4·PF6 is unable to thread through the [21]crown-7 ether in 1·PF6. As this situation is energetically less favourable, there is consequently no driving force for heterodimer formation.
Fig. 5a shows the corresponding ESI mass spectrum recorded after 15 min of equilibration. Clearly, the two homodimers are the dominating species; 122+ appears with ca. 35%, 422+ with ca. 55% intensity. The heterodimer [1·4]2+ is always below 3%. No changes are found when the sample was left to equilibrate for periods as long as a week. This result first of all confirms that heterodimer formation is unfavourable as expected. The low intensity of the heterodimer also indicates that non-specific dimerization does not play a significant role under the experimental conditions applied. The differences of the intensities of the two homodimers point to some differences in the ESI response factors. Nevertheless, a semiquantitative picture is obtained.
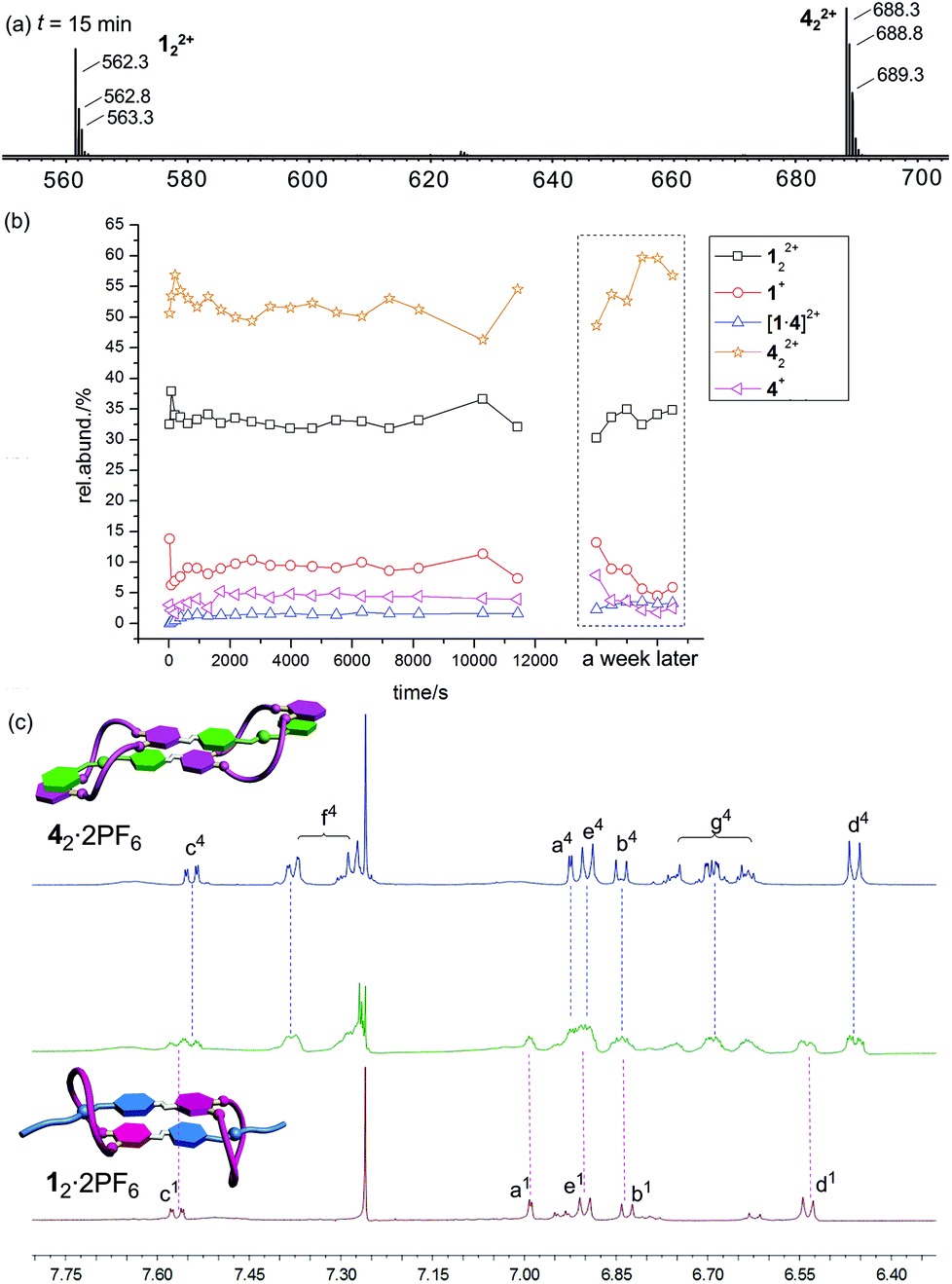 | ||
| Fig. 5 (a) Partial ESI mass spectrum of a 1:1 mixture of 12·2PF6 and 42·2PF6 (100 μM for each monomer) in CHCl3–CH3CN (2:1) recorded after 15 min of equilibration time. (b) More spectra have been recorded after different time intervals. No change is observed even after a week. (c) Partial 1H NMR spectra (500 MHz, CDCl3–CD3CN = 2:1, 10.0 mM, 298 K) of 12·PF6 (bottom row), an equimolar mixture of 12·2PF6 and 42·2PF6 (centre), and 42·2PF6 (top row). | ||
The mass spectrometric results are in agreement with the 1H NMR spectra in Fig. 5c. Although the spectrum of the mixture is somewhat less well-resolved, it is quite clear that this spectrum is a superposition of the two spectra of the homodimers. Full spectra and 1H,1H COSY data are given in the ESI.† In conclusion, both methods are in agreement with the hypothesis that 12·2PF6 and 42·2PF6 do not form heterodimers.
A similar situation is observed for the 2:1 mixture of 2·PF6 and 42·2PF6 that is equimolar in monomer concentration. The heterodimer appears in the mass spectra (ESI†) below 5% intensity accompanied mainly by 2+ and 422+. Due to the larger structural differences between a monomer (2+) and a dimer (422+), larger differences in the ESI response factors can be expected. Indeed, the 2+ ion is much lower in intensity than the 422+ dimer. When judging the intensities, it must however be taken into account that they are proportional to the charge state in Fourier-transform ion-cyclotron-resonance (FTICR) mass spectrometry. Consequently, the 422+ dication intensity is two times higher than that of a singly charged cation of the same abundance. The NMR spectra (ESI†) are again complex, but can be rationalized by a superposition of 2·PF6 and 42·2PF6.
Upon closer inspection, this result might be surprising. As 2·PF6 does not form homodimers, an exchange of 2·PF6 against a monomer in 42·2PF6 to yield the heterodimer would result in the same number of satisfied binding sites and should thus be thermoneutral. Consequently, one might have expected more singly threaded heterodimer and an exchange equilibrium in which 2·PF6, 42·2PF6 and 2·4·2PF6 coexist. However, a doubly threaded, divalent dimer 42·2PF6 may well benefit from chelate cooperativity17 as observed recently for other divalent crown ammonium pseudorotaxanes.9h The open, singly threaded dimer 2·4·2PF6 does of course not profit from such a chelate cooperativity effect.
The 12·2PF6/32·2PF6 (1:1) mixture
Mixing 12·2PF6 and 32·2PF6 in equimolar amounts leads indeed to the expected statistical mixture of 12·2PF6, 1·3·2PF6, and 32·2PF6 as indicated by the appearance of 122+, [1·3]2+ and 322+ in the ESI mass spectrum of the mixture (Fig. 6a). As anticipated, the homodimer dications are superimposed by signals of monomers 1+ and 3+. The intensity ratio of the three dimeric dications is about 3:8:6 (122+:[1·3]2+:322+). If one takes into account the likely differing desolvation energies, this ratio is more or less in agreement with a statistical formation of the three dimeric daisy chains.
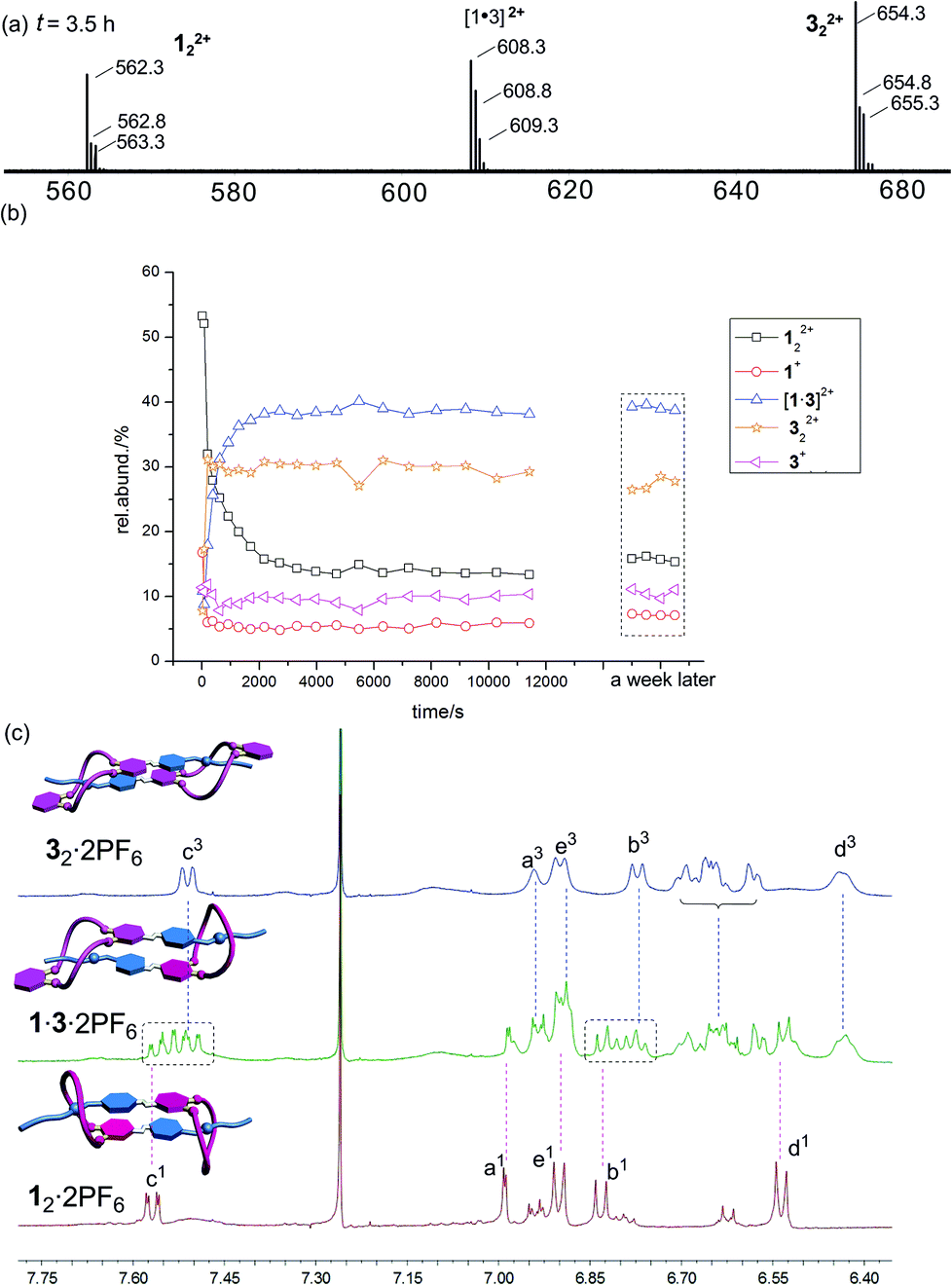 | ||
| Fig. 6 (a) Partial ESI mass spectrum of a 1:1 mixture of 12·2PF6 and 32·2PF6 (100 μM for each monomer) in CHCl3–CH3CN (2:1) recorded after 3.5 h of equilibration time. (b) The equilibration can be followed over time and occurs on a minute time scale. (c) Partial 1H NMR spectra (500 MHz, CDCl3–CD3CN = 2:1, 10.0 mM, 298 K) of 12·2PF6 (bottom row), an equimolar mixture of 12·2PF6 and 32·2PF6 (centre), and 32·2PF6 (top row). | ||
The exchange of monomers between the two homodimers to yield the heterodimeric daisy chain is quite slow and can be followed by ESI mass spectrometry (Fig. 6b). If one takes the development of the heterodimer ion intensity as an example, one arrives at an estimate for the half-life of roughly 230 seconds. The exchange reaction thus proceeds on a minute time scale.
The 1H NMR spectra are very complicated and difficult to interpret – in particular when it comes to quantification of the different dimers present in solution. However, quite clearly, the signals for Hb and Hc of the two compounds in the aromatic region provide evidence that the spectrum of the mixture is more than a mere superposition of the two homodimer spectra. Additional signals are observed, which together with the ESI mass spectrometric results at least provide qualitative evidence for the presence of the homo- and heterodimers in the sample solution.
The 2·PF6/32·2PF6 (2:1) mixture
Finally, the mixture of 2·PF6/32·2PF6 (equimolar in monomer concentration) is expected to self-sort into the heterodimer exclusively. A look at the corresponding ESI mass spectrum (Fig. 7a) confirms the heterodimer to be the most prominent species in the sample solution which again forms on a minute time scale (Fig. 7b). As time passed by, we could see the decrease of the signal of 2+ from 50% to less than 6% and the gradual increase of the intensity of [2·3]2+ (ESI†). Similar ESI mass spectrometric behaviour (higher intensity) were found for 322+ in Fig. 6 and 7, maybe due to its much different desolvation energy from those of other species. Nevertheless, some contribution from 2·PF6 and 32·2PF6 is still observed and one would arrive at the conclusion that self-sorting is imperfect.
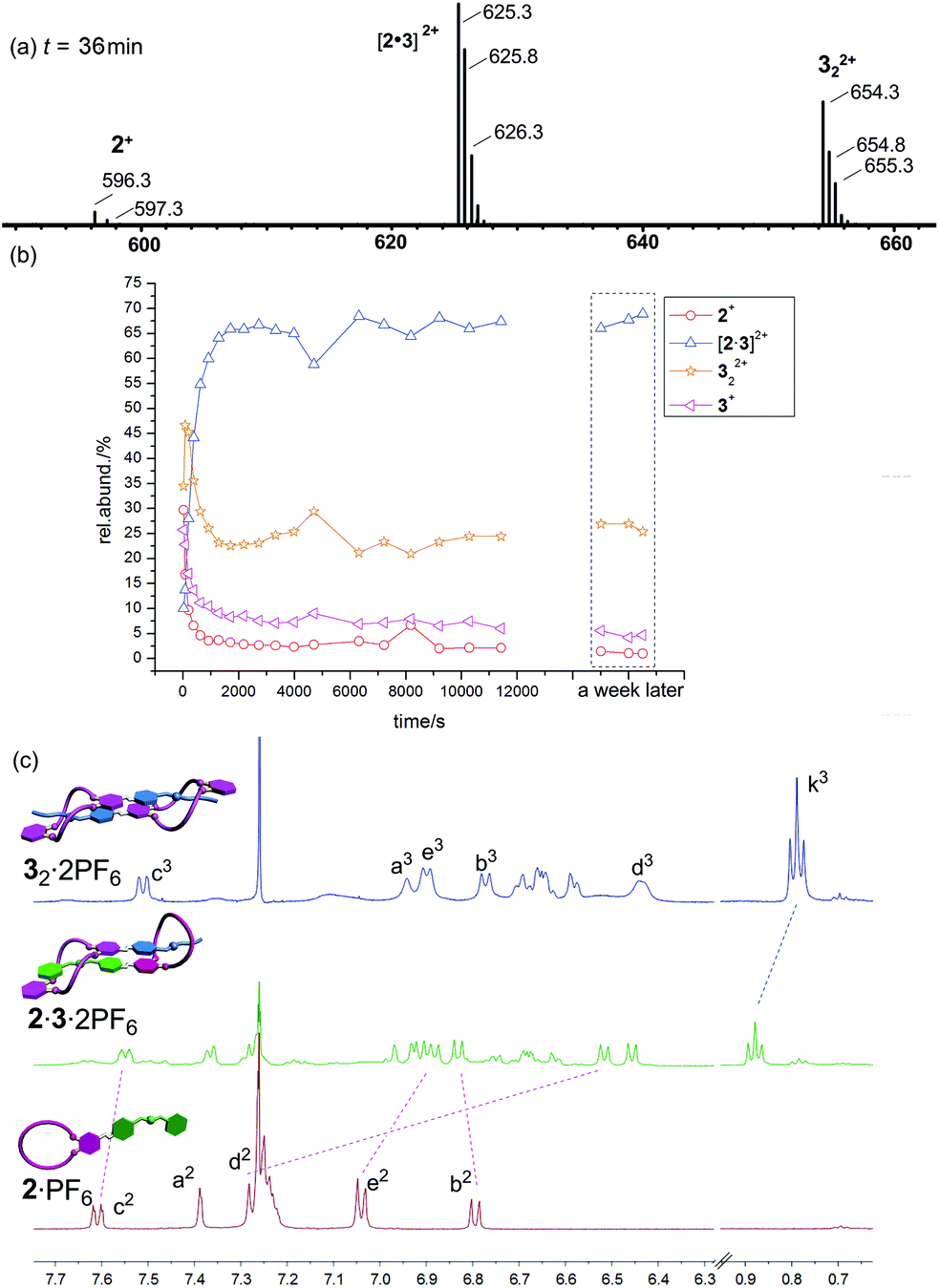 | ||
| Fig. 7 (a) Partial ESI mass spectrum of a 2:1 mixture of 2·PF6 and 32·2PF6 (100 μM for each monomer) in CHCl3–CH3CN (2:1) recorded after 36 min of equilibration time. (b) The self-sorting can be followed over time and occurs on a minute time scale. (c) Partial 1H NMR spectra (500 MHz, CDCl3–CD3CN = 2:1, 10.0 mM, 298 K) of 2·PF6 (bottom row), a 2:1 mixture of 2·PF6 and 32·2PF6 (centre), and 32·2PF6 (top row). | ||
A closer inspection of the NMR spectra of the individual compounds and the mixture of both, however, reveals that self-sorting is almost complete. The 1H NMR spectrum of the mixture exhibits new sets of signals that are not present in the individual parent compounds. In turn, the signals for 2·PF6 and 32·2PF6 are missing in the spectrum of the mixture as seen, for example, for c3, k3 and e2 (Fig. 7c). Such a divergence of the ESI mass spectrometric and NMR spectroscopic results has been observed earlier for other supramolecular self-sorting complexes.3d,e As the NMR (10.0 mM) results reflect the solution situation more directly than ESI mass spectrometry (100 μM), we can therefore conclude that self-sorting quite nicely works for the 2·PF6/32·2PF6 combination.
Experimental section
General
All reagents were commercially available and used as supplied without further purification. Benzo[21]crown-7 carboxylic acid C7-COOH, dibenzo[24]crown-8 carboxylic acid C8-COOH, monomer 1·PF6, and axle precursors 5 and 6 were prepared according to literature procedures.13,141H NMR spectra were collected on a temperature-controlled 500 MHz Bruker AVANCE DMX-500 spectrometer with the deuterated solvent as the lock and the residual undeuterated solvent or TMS as the internal reference. Chemical shifts are reported in ppm and coupling constants in hertz (Hz). 13C NMR spectra were recorded on a Bruker AVANCE DMX-500 spectrometer. The crystal structure data were collected on an Oxford Diffraction Xcalibur Gemini Ultra diffractometer with an Atlas detector. The electrospray-ionization Fourier-transform ion-cyclotron-resonance (ESI-FTICR) mass spectrometric experiments were performed with a Varian/IonSpec QFT-7 FTICR mass spectrometer equipped with a superconducting 7 Tesla magnet and a Micromass Z-spray ESI ion source utilizing a stainless steel capillary with a 0.65 mm inner diameter. The solutions of samples (100 μM in CHCl3–CH3CN 2:1 were introduced into the source with a syringe pump (Harvard Apparatus) at a flow rate of approximately 2–3 μL min−1. The ions were collected in the instrument's accumulation hexapole long enough to obtain useful signal-to-noise ratios, introduced into the FTICR analyzer cell, which was operated at pressures below 10−9 mbar, and detected by a standard excitation and detection sequence. The ionization voltage was set to 3000–3500 V, source and probe temperature were 40 °C. The sample cone voltage was set to 25–30 V.
Syntheses
X-ray crystallographic data
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 11.9860(5), b = 12.2786(7), c = 16.2211(9) Å, α = 103.393(5)°, β = 110.555(5)°, γ = 92.801(4)°, V = 2152.18(19) Å3, Z = 2, Dc = 1.234 g cm−3, T = 140(2) K, μ = 0.140 mm−1, 7859 measured reflections, 3825 independent reflections, 609 parameters, 1729 restraints, F(000) = 840, R1 = 0.1665, wR2 = 0.3278 (all data), R1 = 0.0970, wR2 = 0.2891 [I > 2σ(I)], max. residual density 0.386 e Å−3, and goodness-of-fit (F2) = 1.090. CCDC number: 952285.
339.9(6) Å3, Z = 4, Dc = 1.070 g cm−3, T = 140(2) K, μ = 1.054 mm−1, 13857 measured reflections, 8431 independent reflections, 1040 parameters, 126 restraints, F(000) = 3480, R1 = 0.1859, wR2 = 0.4125 (all data), R1 = 0.1501, wR2 = 0.3841 [I > 2σ(I)], max. residual density 1.172 e Å−3, and goodness-of-fit (F2) = 1.342. CCDC number: 981643.
, a = 11.9860(5), b = 12.2786(7), c = 16.2211(9) Å, α = 103.393(5)°, β = 110.555(5)°, γ = 92.801(4)°, V = 2152.18(19) Å3, Z = 2, Dc = 1.234 g cm−3, T = 140(2) K, μ = 0.140 mm−1, 7859 measured reflections, 3825 independent reflections, 609 parameters, 1729 restraints, F(000) = 840, R1 = 0.1665, wR2 = 0.3278 (all data), R1 = 0.0970, wR2 = 0.2891 [I > 2σ(I)], max. residual density 0.386 e Å−3, and goodness-of-fit (F2) = 1.090. CCDC number: 952285.
339.9(6) Å3, Z = 4, Dc = 1.070 g cm−3, T = 140(2) K, μ = 1.054 mm−1, 13857 measured reflections, 8431 independent reflections, 1040 parameters, 126 restraints, F(000) = 3480, R1 = 0.1859, wR2 = 0.4125 (all data), R1 = 0.1501, wR2 = 0.3841 [I > 2σ(I)], max. residual density 1.172 e Å−3, and goodness-of-fit (F2) = 1.342. CCDC number: 981643.
Conclusions
The results obtained in this study confirm the expected self-sorting behaviour of daisy chain monomers 1·PF6–4·PF6. While the pairs 12·2PF6/42·2PF6 and 2·PF6/42·2PF6 do not form heterodimeric daisy chains, 12·2PF6 and 32·2PF6 yield a more or less statistical mixture. The 2·PF6/32·2PF6 pair instead forms almost exclusively the heterodimeric daisy chain. The present study thus provides evidence that hetero-[c2]daisy chain pseudorotaxanes can be programmed based on the appropriate choice of orthogonal binding motifs. Both motifs used here are very similar and the self-sorting is based on the size complementarity of crown ether size and ammonium axle width.It is, however, this similarity of the two binding motifs which also poses difficulties in the analysis of the solution situation and points to the limits of current routine characterization methods. While X-ray crystallography and NMR experiments are quite straightforward, when it comes to the characterization of the monomers and homodimers, the determination of solution concentrations in mixtures of them is not trivial – even though the molecules under study are structurally not very complicated.
Finally, a combination of ESI mass spectrometry and NMR spectroscopy provides semi-quantitative insight into the self-sorting behaviour as well as a rough estimate on the equilibration time scale. Nevertheless, to know the self-sorting behaviour is a key issue for future studies. For example, 2·PF6 and 3·PF6 can now be equipped with two different functional groups. They can then be brought into proximity to each other, when the heterodimer daisy chain forms – thus providing complexes with two functionalities which might be capable of cooperating with each other e.g. in organocatalysis. The two point attachment between the two monomers provides the basis for better geometrical control over the complex formed than a single-site recognition between the two partners. Furthermore, the exclusive formation of the heterodimer maximizes the concentration of complexes that bear both functional groups and therefore maximizes complex activity. The use of the crown ether/ammonium ion binding motifs renders the complexes switchable by (de)protonation. Consequently, the cooperation of the two functional groups could be switched on and off.
Acknowledgements
F.H. thanks the financial support from the National Basic Research Program (2013CB834502) and the National Natural Science Foundation of China (21125417). C.A.S. greatly appreciates funding by the Deutsche Forschungsgemeinschaft (SFB 765). We thank Dr Henrik Winkler for orienting measurements. B.Z. thanks the Center for International Cooperation of Freie Universität Berlin for a Ph.D. exchange fellowship.Notes and references
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312–1319 CAS.
- For reviews, see: (a) M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed; (b) M. L. Saha and M. Schmittel, Org. Biomol. Chem., 2012, 10, 4651–4684 RSC.
- Selected examples: (a) Y. Ma, S. V. Kolotuchin and S. C. Zimmerman, J. Am. Chem. Soc., 2002, 124, 13757–13769 CrossRef CAS PubMed; (b) A. Wu and L. Isaacs, J. Am. Chem. Soc., 2003, 125, 4831–4835 CrossRef CAS PubMed; (c) E. S. Barrett, T. J. Dale and J. Rebek Jr., J. Am. Chem. Soc., 2008, 130, 2344–2350 CrossRef CAS PubMed; (d) Y. Rudzevich, V. Rudzevich, F. Klautzsch, C. A. Schalley and V. Böhmer, Angew. Chem., Int. Ed., 2009, 48, 3867–3871 CrossRef CAS PubMed; (e) C. Talotta, C. Gaeta, Z. Qi, C. A. Schalley and P. Neri, Angew. Chem., Int. Ed., 2013, 52, 7437–7441 CrossRef CAS PubMed.
- Selected examples: (a) P. N. Taylor and H. L. Anderson, J. Am. Chem. Soc., 1999, 121, 11538–11545 CrossRef CAS; (b) J.-M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS PubMed; (c) I.-W. Hwang, T. Kamada, T. K. Ahn, D. M. Ko, T. Nakamura, A. Tsuda, A. Osuka and D. Kim, J. Am. Chem. Soc., 2004, 126, 16187–16198 CrossRef CAS PubMed; (d) K.-W. Chi, C. Addicott, A. M. Arif and P. J. Stang, J. Am. Chem. Soc., 2004, 126, 16569–16574 CrossRef CAS PubMed; (e) B. H. Northrop, Y.-R. Zheng, K.-W. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554–1563 CrossRef CAS PubMed; (f) Y. R. Zheng;, H. B. Yang, K. Ghosh, L. Zhao and P. J. Stang, Chem. – Eur. J., 2009, 15, 7203–7214 CrossRef PubMed; (g) S. Ulrich and J.-M. Lehn, J. Am. Chem. Soc., 2009, 131, 5546–5549 CrossRef CAS PubMed; (h) S. Ulrich and J.-M. Lehn, Chem. – Eur. J., 2009, 15, 5640–5645 CrossRef CAS PubMed; (i) K. Mahata and M. Schmittel, J. Am. Chem. Soc., 2009, 131, 16544–16554 CrossRef CAS PubMed; (j) T. Murase, S. Horiuchi and M. Fujita, J. Am. Chem. Soc., 2010, 132, 7864–7865 CrossRef CAS PubMed; (k) Y. Yamauchi, M. Yoshizawa, M. Akita and M. Fujita, J. Am. Chem. Soc., 2010, 132, 960–966 CrossRef CAS PubMed; (l) K. Mahata, M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2010, 132, 15933–15935 CrossRef CAS PubMed; (m) M. Schmittel and K. Mahata, Chem. Commun., 2010, 46, 4163–4165 RSC; (n) M. M. J. Smulders, A. Jiménez and J. R. Nitschke, Angew. Chem., Int. Ed., 2012, 51, 6681–6685 CrossRef CAS PubMed; (o) A. S. Singh and S.-S. Sun, Chem. Commun., 2012, 48, 7392–7394 RSC; (p) M. L. Saha, S. Pramanik and M. Schmittel, Chem. Commun., 2012, 48, 9459–9461 RSC.
- Selected examples: (a) A. D. Shaller, W. Wang, H. Gan and A. D. Q. Li, Angew. Chem., Int. Ed., 2008, 47, 7705–7709 CrossRef CAS PubMed; (b) S. Ghosh, A. Wu, J. C. Fettinger, P. Y. Zavalij and L. Isaacs, J. Org. Chem., 2008, 73, 5915–5925 CrossRef CAS PubMed; (c) S. Ghosh, X.-Q. Li, V. Stepanenko and F. Würthner, Chem. – Eur. J., 2008, 14, 11343–11357 CrossRef CAS PubMed; (d) M. M. Safont-Sempere, P. Osswald, M. Stolte, M. Grüne, M. Renz, M. Kaupp, K. Radacki, H. Braunschweig and F. Würthner, J. Am. Chem. Soc., 2011, 133, 9580–9591 CrossRef CAS PubMed.
- Selected examples: (a) N. Tomimasu, A. Kanaya, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2009, 131, 12339–12343 CrossRef CAS PubMed; (b) W. Jiang, Q. Wang, I. Linder, F. Klautzsch and C. A. Schalley, Chem. – Eur. J., 2011, 17, 2344–2348 CrossRef CAS PubMed.
- (a) P. Mukhopadhyay, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2006, 128, 14093–14102 CrossRef CAS PubMed; (b) P. Mukhopadhyay, A. Wu and L. Isaacs, J. Org. Chem., 2004, 69, 6157–6164 CrossRef CAS PubMed.
- (a) P. R. Ashton, E. J. T. Chrystal, P. T. Glink, S. Menzer, C. Schiavo, N. Spencer, J. F. Stoddart, P. A. Tasker, A. J. P. White and D. J. Williams, Chem. – Eur. J., 1996, 2, 709–728 CrossRef CAS; (b) N. Yamaguchi, L. M. Hamilton and H. W. Gibson, Angew. Chem., Int. Ed., 1998, 37, 3275–3279 CrossRef CAS; (c) P. R. Ashton, I. Baxter, M. C. T. Fyfe, F. M. Raymo, N. Spencer, J. F. Stoddart, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 2297–2307 CrossRef CAS; (d) T. Clifford, A. Abushamleh and D. H. Busch, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4830–4836 CrossRef CAS PubMed; (e) F. Huang, K. A. Switek, L. N. Zakharov, F. R. Fronczek, C. Slebodnick, M. Lam, J. A. Golen, W. S. Bryant, P. E. Mason, A. L. Rheingold, M. Ashraf-Khorassani and H. W. Gibson, J. Org. Chem., 2005, 70, 3231–3241 CrossRef CAS PubMed; (f) Z. Niu, F. Huang and H. W. Gibson, J. Am. Chem. Soc., 2011, 133, 2836–2839 CrossRef CAS PubMed.
- (a) F. Wang, C. Han, C. He, Q. Zhou, J. Zhang, C. Wang, N. Li and F. Huang, J. Am. Chem. Soc., 2008, 130, 11254–11255 CrossRef CAS PubMed; (b) W. Jiang, H. D. F. Winkler and C. A. Schalley, J. Am. Chem. Soc., 2008, 130, 13852–13853 CrossRef CAS PubMed; (c) W. Jiang and C. A. Schalley, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10425–10429 CrossRef CAS PubMed; (d) F. Wang, B. Zheng, K. Zhu, Q. Zhou, C. Zhai, S. Li, N. Li and F. Huang, Chem. Commun., 2009, 4375–4377 RSC; (e) W. Jiang, A. Schäfer, P. C. Mohr and C. A. Schalley, J. Am. Chem. Soc., 2010, 132, 2309–2320 CrossRef CAS PubMed; (f) W. Jiang and C. A. Schalley, J. Mass Spectrom., 2010, 45, 788–798 CrossRef CAS PubMed; (g) W. Jiang, D. Sattler, K. Rissanen and C. A. Schalley, Org. Lett., 2011, 13, 4502–4505 CrossRef CAS PubMed; (h) W. Jiang, K. Nowosinski, N. L. Löw, E. V. Dzyuba, F. Klautzsch, A. Schäfer, J. Huuskonen, K. Rissanen and C. A. Schalley, J. Am. Chem. Soc., 2012, 134, 1860–1868 CrossRef CAS PubMed; (i) S. Dong, B. Zheng, M. Zhang, X. Yan, X. Ding, Y. Yu and F. Huang, Macromolecules, 2012, 45, 9070–9075 CrossRef CAS; (j) S. Dong, X. Yan, B. Zheng, J. Chen, X. Ding, Y. Yu, D. Xu, M. Zhang and F. Huang, Chem. – Eur. J., 2012, 18, 4195–4199 CrossRef CAS PubMed.
- (a) P. R. Ashton, P. J. Campbell, P. T. Glink, D. Philp, N. Spencer, J. F. Stoddart, E. J. T. Chrystal, S. Menzer, D. J. Williams and P. A. Tasker, Angew. Chem., Int. Ed. Engl., 1995, 34, 1865–1869 CrossRef CAS; (b) C. Zhang, S. Li, J. Zhang, K. Zhu, N. Li and F. Huang, Org. Lett., 2007, 9, 5553–5556 CrossRef CAS PubMed.
- Reviews: (a) L. Fang, M. A. Olson, D. Benitez, E. Tkatchouk, W. A. Goddard III and J. F. Stoddart, Chem. Soc. Rev., 2010, 39, 17–29 RSC; (b) J. Rotzler and M. Mayor, Chem. Soc. Rev., 2013, 42, 44–62 RSC; (c) B. Zheng, F. Wang, S. Dong and F. Huang, Chem. Soc. Rev., 2012, 41, 1621–1636 RSC. Selected examples: (d) P. R. Ashton, I. Baxter, S. J. Cantrill, M. C. T. Fyfe, P. T. Glink, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 1294–1297 CrossRef CAS; (e) S. J. Cantrill, G. J. Youn, J. F. Stoddart and D. J. Williams, J. Org. Chem., 2001, 66, 6857–6872 CrossRef CAS PubMed; (f) D. G. Amirsakis, A. M. Elizarov, M. A. Garcia-Garibay, P. T. Glink, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 2003, 42, 1126–1132 CrossRef CAS PubMed; (g) M. P. L. Werts, M. van den Boogaard, G. M. Tsivgoulis and G. Hadziioannou, Macromolecules, 2003, 36, 7004–7013 CrossRef CAS; (h) H. W. Gibson, N. Yamaguchi, Z. Niu, J. W. Jones, C. Slebodnick, A. L. Rheingold and L. N. Zakharov, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 975–985 CrossRef CAS; (i) L. Cao and L. Isaacs, Org. Lett., 2012, 14, 3072–3075 CrossRef CAS PubMed; (j) Z. Zhang, G. Yu, C. Han, J. Liu, X. Ding, Y. Yu and F. Huang, Org. Lett., 2011, 13, 4818–4821 CrossRef CAS PubMed; (k) Z. Zhang, C. Han, G. Yu and F. Huang, Chem. Sci., 2012, 3, 3026–3031 RSC.
- Review: (a) J.-P. Collin, C. Dietrich-Buchecker, P. Gaviña, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477–487 CrossRef CAS PubMed; examples: (b) C. Romuald, E. Busseron and F. Coutrot, J. Org. Chem., 2010, 75, 6516–6531 CrossRef CAS PubMed; (c) G. Du, E. Moulin, N. Jouault, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2012, 51, 12504–12508 CrossRef CAS PubMed; (d) X. Yan, B. Zheng and F. Huang, Polym. Chem., 2013, 4, 2395–2399 RSC.
- B. Zheng, M. Zhang, S. Dong, J. Liu and F. Huang, Org. Lett., 2012, 14, 306–309 CrossRef CAS PubMed.
- (a) D.-J. Feng, X.-Q. Li, X.-Z. Wang, X.-K. Jiang and Z.-T. Li, Tetrahedron, 2004, 60, 6137–6144 CrossRef CAS PubMed; (b) L. Wu, Y.-M. He and Q.-H. Fan, Adv. Synth. Catal., 2011, 353, 2915–2919 CrossRef CAS.
- S. Di Stefano, J. Phys. Org. Chem., 2010, 23, 797–805 CrossRef CAS.
- E. Leize, A. Jaffrezic and A. Van Dorsselaer, J. Mass Spectrom., 1996, 31, 537–544 CrossRef CAS.
- For reviews on multivalency, see: (a) M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef; (b) A. Mulder, J. Huskens and D. N. Reinhoudt, Org. Biomol. Chem., 2004, 2, 3409–3424 RSC; (c) J. D. Badjic, A. Nelson, S. J. Cantrill, W. B. Turnbull and J. F. Stoddart, Acc. Chem. Res., 2005, 38, 723–732 CrossRef CAS PubMed; (d) C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed. For reviews on the thermodynamic analysis of chelate cooperativity in supramolecular complexes, see: (e) G. Ercolani, C. Piguet, M. Borkovec and J. Hamacek, J. Phys. Chem. B, 2007, 111, 12195–12203 CrossRef CAS PubMed; (f) C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488–7499 CrossRef CAS PubMed; (g) G. Ercolani and L. Schiaffino, Angew. Chem., Int. Ed., 2011, 50, 1762–1768 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional NMR and MS data, ellipsoid plots of new crystal structures. CCDC 952285 and 981643. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00064a |
| This journal is © the Partner Organisations 2014 |