Efficient synthesis of a library of heparin tri- and tetrasaccharides relevant to the substrate of heparanase†
Peng
Xu
,
Weichang
Xu
,
Yuanwei
Dai
,
You
Yang
and
Biao
Yu
*
State Key Laboratory of Bio-organic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: byu@mail.sioc.ac.cn
First published on 21st March 2014
Abstract
The glycosylation reaction for construction of the challenging α-GlcN–(1→4)-GlcA/IdoA linkages has been investigated carefully. A standard protocol was thus fixed that employed 2-azido-glucopyranosyl N-phenyl trifluoroacetimidates as donors, TMSOTf as a catalyst, toluene as a solvent, and −30 °C as the working temperature. With this protocol, a variety of mono- and disaccharide donors and acceptors were condensed reliably to provide the corresponding coupled tri- and tetrasaccharides in satisfactory yields and α-selectivity, whereas a remote protecting group or a sugar unit in either the donor or the acceptor did affect considerably the outcome. The resulting tri- and tetrasaccharides bearing orthogonal protecting groups were then converted efficiently into the corresponding heparin tri- and tetrasaccharides via a robust approach involving saponification, O-sulfation, azide reduction, N-sulfation/N-acetylation, and global debenzylation. These heparin tri- and tetrasaccharides are structurally relevant to ΔHexA(2S)–GlcN(NS,6S)–GlcA–GlcN(NS,6S), a reported substrate of heparanase, and therefore could be exploited to examine the substrate specificity of this important enzyme.
Introduction
Heparin (HP) and heparan sulfate (HS) are highly sulfated linear polysaccharides that represent the most complex members of the glycosaminoglycan (GAG) family.1 HP, found mainly on mast cells, consists of alternating (1→4)-linked uronic acid (either α-L-iduronic acid (IdoA) or β-D-glucuronic acid (GlcA)) and α-D-glucosamine (GlcN) units.1,2 HS, which is ubiquitously distributed on the surface of animal cells and in the extracellular matrix (ECM), shares the same backbone with HP but carries less sulfate groups and a lower percentage of the IdoA residue.1,3–5 The interaction of HP/HS and proteins plays a key role in numerous physiological processes such as blood coagulation, angiogenesis, lipid metabolism, tumor metastasis, growth factor binding, cell adhesion, inflammatory response, and bacterial and viral infections.1,3,6–8 These functions of HP/HS are to a large extent determined by the saccharide sequence, chain length, and sulfation patterns.9 In particular, an HP pentasaccharide specifically binding to antithrombin III has been developed as an anticoagulant drug (Fondaparinux).2,9 Nevertheless, the structure–activity relationships (SAR) of HP/HS in most HP/HS-mediated biological events are still poorly understood.Heparanase is a mammalian endo-β-D-glucuronidase that degrades HP/HS at specific sites, so as to reduce the integrity of ECM and basement membranes and release the HS-bound growth factors.10,11 Overexpression of heparanase is closely associated with the disease state of tumor progression and metastasis.12 Thus, heparanase has become an important target for developing anticancer drugs.13 Previous studies on the substrate specificity of the human heparanase revealed the tetrasaccharide ΔHexA(2S)–GlcN(NS,6S)–GlcA–GlcN(NS,6S) as an ideal sequence.14 An optimal substrate derivative could be exploited to establish an efficient assay of the heparanase activity, thus facilitating the studies on the function of this important enzyme and the development of inhibitors.15
Chemical (and chemoenzymatic) syntheses of HP/HS have been studied intensively;5,9,16–19 various protocols/strategies for the preparation of building blocks, glycosylation, and protecting group manipulation have been explored, such as modular assembly,17c,j,18h,l,p one-pot method,18e,i pre-sulfated assembly,18jde novo strategy,18d solid-phase synthesis,17e,18q and diversity-oriented synthesis.18b,k,m However, the α-selective formation of the GlcN–(1→4)-GlcA/IdoA glycosidic linkages remains elusive; the difficult-to-separate or inseparable β anomers often result in various amounts.20 2-Azide (mostly),21 2N,3O-oxazolidinone,22 and N-p-methoxybenzylidene23 are employed as amino protecting groups in the GlcN donors to ensure a non-participating effect on the glycosylation, nevertheless all other reaction parameters including the protecting patterns of both the donor and the acceptor, the anomeric leaving group, the promoter, the solvent, the temperature, and the additive also affect seriously the coupling yield and α-selectivity.20,24 Herein, we report the synthesis of a library of tri- and tetrasaccharides (1–20) structurally relevant to ΔHexA(2S)–GlcN(NS,6S)–GlcA–GlcN(NS,6S) (Fig. 1), whereas the α-selective glycosylation with 2-azido-glucopyranosyl donors to construct the GlcN–(1→4)-GlcA/IdoA linkages has been carefully studied.
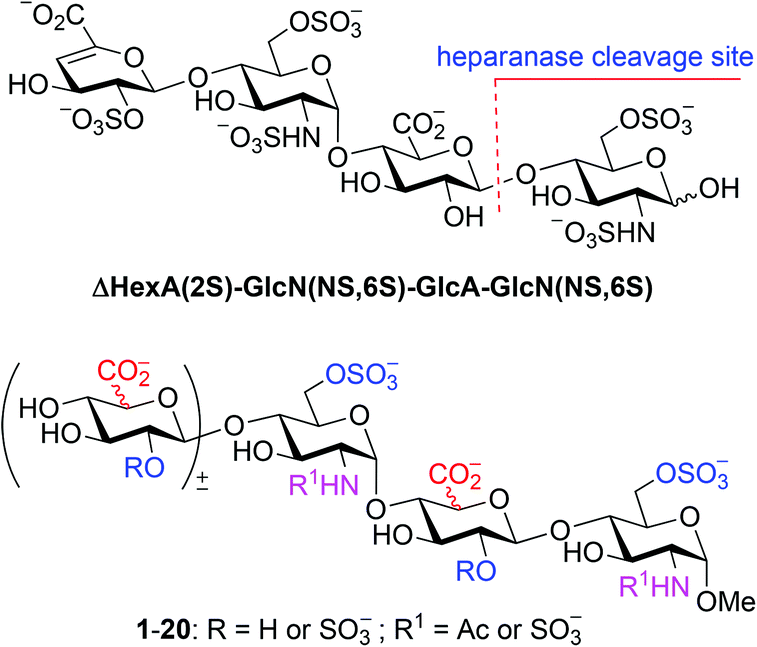 | ||
| Fig. 1 An optimal substrate of heparanase and a library of the structurally relevant oligosaccharides (1–20). | ||
Results and discussion
Synthetic strategy
The GlcA/IdoA units in HP/HS are mostly decorated with 2-O-sulfation, whereas the GlcN units are typically substituted with 2-N-acetylation/sulfation and 6-O-sulfation.25,26 A judicious choice of a set of glycosyl building blocks installed with orthogonal protecting groups is crucial for the efficient construction of a library of HS oligosaccharides.9,16–18 To synthesize tri- and tetrasaccharides 1–20 in a combinatorial manner, glucose (Glc) building blocks 21 and 22, an idose (Ido) building block 23, and GlcN building blocks 24–28 were designed to meet the requirements of stereoselective glycosylation, chain elongation, and regioselective sulfation (Fig. 2). As such, the 4,6-hydroxyl groups in 21–23 were protected as 4,6-O-benzylidene acetals that would be oxidized to carboxylic acids followed by blocking with methyl groups at the C6 position with the 4-OH being free for chain elongation. The anomeric tert-butyldimethylsilyl (TBS) groups in GlcN 25 and 26 could be selectively removed with tetrabutylammonium fluoride (TBAF) for further conversion into glycosyl leaving groups. Moreover, the benzoyl (Bz), levulinate (Lev),27 and p-methoxybenzyl (PMB) groups were selected to protect the hydroxyl groups destined for sulfation, while the benzyl (Bn) group was employed as the permanent protecting group. The amine group was masked as an azide that could be readily reduced into free amine for N-acetylation or N-sulfation at a late stage.20 | ||
| Fig. 2 Monosaccharide building blocks 21–28. | ||
Couplings of Glc/Ido 21–23 with GlcN 25–28 would provide a range of Glc/Ido–(1→4)-GlcN disaccharides, where the 2-ester groups (O-Bz or O-Lev) would secure the formation of the required 1,2-trans glycosidic linkages via the aid of neighboring group participation.28 The resulting Glc/Ido–GlcN disaccharides could be readily converted into GlcA/IdoA–GlcN donors and acceptors.29 The α-selective [2 + 2] glycosidic coupling to construct the GlcN–(1→4)-GlcA/IdoA linkages could then be explored. Employing an optimal set of standard conditions, we could assemble all the building blocks into oligosaccharides. Conventional operation of the protecting groups and timely sulfation would lead to the desired tri- and tetrasaccharides (1–20).
Preparation of the disaccharide building blocks
The eight differentially protected monosaccharide building blocks 21–28 were readily prepared in large quantities adopting the literature procedures.18f,30,31 Glycosylation of Glc trichloroacetimidate3221 with GlcN 25 and 26 under the catalysis of TMSOTf (0.05 equiv.) provided β-(1→4) linked disaccharides 29 and 30 in >90% yields (Scheme 1). Treatment of 29 and 30 with BH3·THF and Bu2BOTf led to regioselective opening of the 4′,6′-O-benzylidene acetal, affording the disaccharides with free 6-hydroxyl groups at the Glc residues.33 It should be noted that other BH3·THF-mediated reduction on 29 and 30 using Cu(OTf)2, Sc(OTf)3, or TMSOTf as Lewis acid failed to give the desired disaccharides in good yield.34 The resulting 6-hydroxyl groups were then oxidized with 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) in the presence of [bis(acetoxy)iodo]benzene (BAIB),29 and subsequent methylation of the nascent carboxylic acids afforded GlcA–GlcN disaccharides 31 and 32 in 75% and 77% yields, respectively, over three steps. Removal of the anomeric TBS groups in 31 and 32 with TBAF and acetic acid gave the corresponding lactols, which were readily converted into glycosyl o-cyclopropylethynylbenzoate3533, trichloroacetimidate 34, and N-phenyl trifluoroacetimidate3635 and 36, respectively.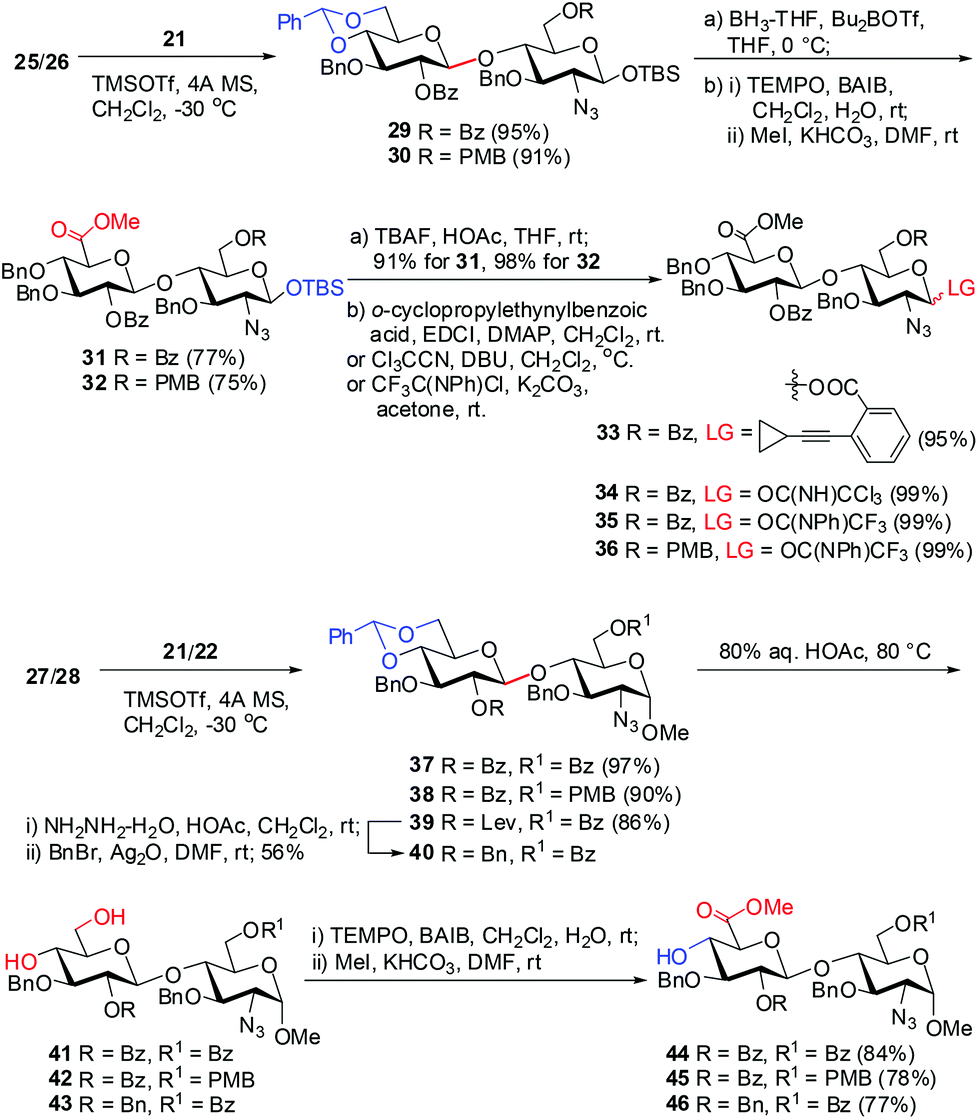 | ||
| Scheme 1 Preparation of the GlcA–GlcN disaccharide donors 33–36 and acceptors 44–46. | ||
On the other hand, TMSOTf-catalyzed glycosylation of Glc imidates 21/22 with GlcN derivatives 27/28 was performed to provide disaccharides 37–39 in high yields (86–97%), whereas the 2-O-Bz or -Lev group in the Glc donors participated in the glycosylation to secure the exclusive formation of β-linkage. The Lev group in 39 was then replaced by the permanent Bn protecting group. Acidic cleavage of the benzylidene acetals in 37, 38, and 40 with aqueous acetic acid at 80 °C followed by TEMPO oxidation and subsequent methylation furnished disaccharide acceptors 44–46 in 77–84% yields over three steps.
Similarly, glycosylation of GlcN derivatives 25 and 27 with Ido trichloroacetimidate 23 proceeded smoothly under the catalysis of TMSOTf (0.1 equiv.), leading to Ido–GlcN disaccharides 47 and 50 in excellent yields (Scheme 2). Following the above procedures for the conversion of 29→35 and 37→44, Ido–GlcN 47 and 50 were easily transformed into IdoA–GlcN 49 and 52 in good yields.
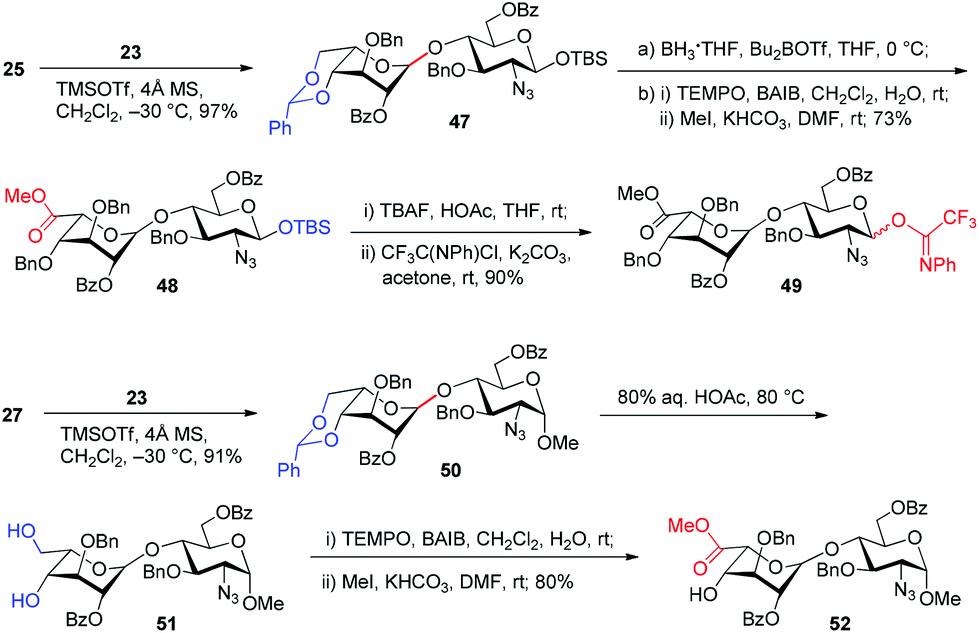 | ||
| Scheme 2 Preparation of the IdoA–GlcN disaccharide donor 49 and acceptor 52. | ||
Investigation of the α-selective glycosylation to construct the GlcN–(1→4)-GlcA/IdoA linkages
With GlcA–GlcN derivative 44 as an acceptor, we examined the [2 + 2] glycosylation with GlcA–GlcN ortho-alkynylbenzoate 33, trichloroacetimidate 34, and N-phenyltrifluoroacetimidate 35 as donors. The representative results are listed in Table 1. The coupling of 44, which bears a hindered 4′-OH, with ortho-alkynylbenzoate 33 proceeded sluggishly under the catalysis of PPh3AuOTf (0.1 equiv.) at room temperature. With toluene as the solvent,37 the reaction led to the coupled tetrasaccharide 53 in a moderate 41% yield (α/β = 4.4) (entry 1). CH2Cl2 or ClCH2CH2Cl was proved to be the worse solvent for this coupling; the reaction hardly took place, with the α-anomer 53α being isolated in only ∼10% yield after ∼8 h (entry 2). In these reactions, the donor and acceptor could be largely recovered.|
|
|||||
|---|---|---|---|---|---|
| Entry | Donor | Promotera | Solvent | Temp. | Yield (α/β ratio) |
| a 0.1 equiv. was used. b 2-Bromothiophene (12 equiv.) was added as an additive. | |||||
| 1 | 33 | PPh3AuOTf | Toluene | rt | 41% (α/β = 4.4) |
| 2 | PPh3AuOTf | CH2Cl2 or ClCH2CH2Cl | rt | ∼10% (α only) | |
| 3 | 34 | TMSOTf | CH2Cl2 or THF | −30 °C | Trace |
| 4 | TBSOTf | Toluene | −30 °C | 62% (α/β = 3) | |
| 5 | 35 | TMSOTf | THF | −30 °C | trace |
| 6 | TMSOTf | Et2O | −30 °C | 54% (α/β = 10) | |
| 7 | TMSOTf | CH2Cl2 | −30 °C | 39% (α/β = 4.2) | |
| 8 | TBSOTf | CH2Cl2 | −30 °C | 34% (α/β = 6) | |
| 9 | TBSOTf | Toluene | −30 °C | 58% (α/β = 2.5) | |
| 10 | TMSOTf | Toluene | −30 °C | 81% (α/β = 3.8) | |
| 11b | TMSOTf | Toluene | −30 °C | 72% (α/β = 6) | |

The coupling of GlcA–GlcN trichloroacetimidates with GlcA–GlcN acceptors was reported to provide the coupled tetrasaccharides in moderate to good yields with varied α-selectivity.18h,38 The condensation of 34 and 44 in the presence of TMSOTf (0.1 equiv.) in CH2Cl2 or THF38 at −30 °C proceeded sluggishly, thereby leading to the formation of 53 in only trace amounts (entry 3). This unexpected result implies the mismatch of these two coupling partners.18l,o,38 Interestingly, when TBSOTf was employed as the catalyst and toluene as the solvent, the reaction of 34 and 44 delivered tetrasaccharide 53 in a good 62% yield with moderate stereoselectivity (α/β = 3) (entry 4). The anomeric configuration of tetrasaccharide 53 was confirmed by comparison of the chemical shifts of the anomeric carbons of 53α (δ 97.2, 98.6, 101.0, 101.4 ppm) with those of 53β (δ 98.6, 101.1, 101.4, 101.9 ppm).31 Due to the observation of the corresponding glycosyl trichloroacetamide which was derived from trichloroacetimidate 34, we then focused our attention on the N-phenyl trifluoroacetimidate donor 35 for further optimization of the present [2 + 2] glycosylation reaction.18f
The coupling of trifluoroacetimidate 35 with 44 in THF (0.1 equiv. TMSOTf, −30 °C) led to a trace amount of the coupled tetrasaccharide 53 (entry 5). Nevertheless, with Et2O as the solvent the coupling provided 53 in 54% yield and an excellent α selectivity (α/β = 10) (entry 6). Unfortunately, this reaction took two days due to the poor solubility of 35 and 44 in ether. Replacing the solvent with CH2Cl2, the reaction under the catalysis of either TMSOTf or TBSOTf led to lower yield of the tetrasaccharide (<40%) and α selectivity (α/β = 4.2 and 6, respectively; entries 7 and 8). Toluene was again proved to be a superior solvent in the present glycosylation, and the coupling of 35 and 44 catalyzed by TBSOTf provided 53 in 58% yield (α/β = 2.5) in ∼2 h (entry 9). The yield of 53 was improved significantly to 81% when TMSOTf was used as the catalyst, although the α selectivity was still moderate (α/β = 3.8; entry 10). The addition of 2-bromothiophene (12 equiv.) indeed improved the α-selectivity (α/β = 6), whereas the yield decreased slightly (72%; entry 11).39
Taking all together the yield, α selectivity, reaction time, and ease of purification, we decided to utilize glycosyl N-phenyl trifluoroacetimidate as a donor, TMSOTf as a catalyst, toluene as a solvent, and −30 °C as the working temperature to further explore the [2 + 2] coupling of the disaccharide building blocks (Scheme 3). Under these fixed conditions, the condensation of GlcA/IdoA–GlcN trifluoroacetimidates 35, 36, and 49 with GlcA/IdoA–GlcN acceptors 44–46 and 52 provided the corresponding tetrasaccharides 54–59 in good yields (58–77%) and satisfactory α selectively (from α/β = 3.2 to α only). In comparison, the glycosylation with IdoA–GlcN donor (49) showed a higher α-selectivity (α/β = 7.5 to α only vs. α/β = 3.2–6.4) than those with GlcA–GlcN donors (35 and 36), irrespective of the acceptors (44–46 and 52). When GlcA–GlcN 35 was used as a donor, the glycosylation with IdoA–GlcN acceptor 52 and GlcA–GlcN acceptor 44, which bear a similar protecting group pattern displayed a similar α-selectivity (α/β = 3.2 vs. α/β = 3.8). The acceptor bearing the 2′-O-Bn group (i.e., 46) gave a slightly higher α-selectivity (α/β = 4.9 vs. 3.2–3.8) than acceptors bearing the 2′-O-Bz group (i.e., 44 and 52) upon coupling with donor 35. In addition, IdoA/GlcN acceptor 52 exhibited a slightly better α-selectivity (α only vs. α/β = 7.5–10.5) than GlcA–GlcN acceptors 44 and 46 when IdoA–GlcN 49 was employed as a donor.
 | ||
| Scheme 3 The [2 + 2] couplings of GlcA/IdoA–GlcN donors 35, 36, and 49 with GlcA/IdoA–GlcN acceptors 44–46 and 52 under the fixed conditions (0.1 equiv. TMSOTf, 5 Å MS, toluene, −30 °C). | ||
To further examine the influence of acceptors on the α-selectivity in the present glycosylation, we carried out the relevant couplings with GlcN trifluoroacetimidate 24 as a simplified donor (Scheme 4). Interestingly, IdoA–GlcN acceptor 52 displayed a lower α-selectivity (2.2 vs. 5.7 to α only) than GlcA–GlcN acceptors 44 and 46 in the [1 + 2] coupling. However, when GlcA 63 and IdoA 65 were employed as acceptors, a reverse α-preference was found in the coupling with 24 compared to the [1 + 2] coupling. These results are consistent with the previous findings.17c,j,18o Specifically, the coupling of GlcN 24 and IdoA acceptor 65 furnished exclusively the α-disaccharide 66 in 90% yield, while glycosylation of GlcN 24 with GlcA acceptor 63 led to disaccharide 66 in only 48% yield with a moderate α/β ratio (α/β = 5.5) (Scheme 4).40 In general, the α-selectivity in the glycosylation with GlcN donors is to a large extent influenced by the additional sugar units in both the donors and acceptors.17g,38,41
 | ||
| Scheme 4 [1 + 2] and [1 + 1] glycosylation with GlcN donor 24 under the fixed conditions (0.1 equiv. TMSOTf, 5 Å MS, toluene, −30 °C). | ||
Syntheses of a library of the HS tri- and tetrasaccharides
The orthogonally protected tri- and tetrasaccharides 53–62 were converted readily into the corresponding HS tri- and tetrasaccharides (1–20) bearing the desired sequence and sulfation patterns. With the conversion of tetrasaccharide 53α to HS tetrasaccharides 1 and 2 as an example, this final elaboration involves six steps (Scheme 5). (1) The methyl esters were cleaved with LiOH and H2O2 in THF;42 (2) the remaining benzoyl groups were removed completely with KOH in methanol; (3) the resulting hydroxyl groups were sulfated with SO3·pyridine; (4) the azides were reduced with Me3P in the presence of NaOH;43 (5) the resulting amines were N-acetylated with acetic anhydride or N-sulfated with SO3·pyridine; and (6) the benzyl groups were finally removed via hydrogenolysis over Pd(OH)2/C in a mixture of methanol and water. For the conversion of 56α to 5/6, an additional step was carried out at the beginning to remove the PMB groups with dichlorodicyanobenzoquinone (DDQ).44 All these steps are high-yielding and robust, and were thus applied reliably to the synthesis of all the desired HS tri- and tetrasaccharides (1–20) with the overall yields of 35–78% (Scheme 6).31 The NMR spectra, especially the easily diagnostic chemical shifts of the anomeric carbons, of the synthetic 1–20 were found to be virtually identical to those reported for the analogous oligosaccharides in the literature.16–18 The sulfation numbers were well confirmed by ESI-MS analyses.31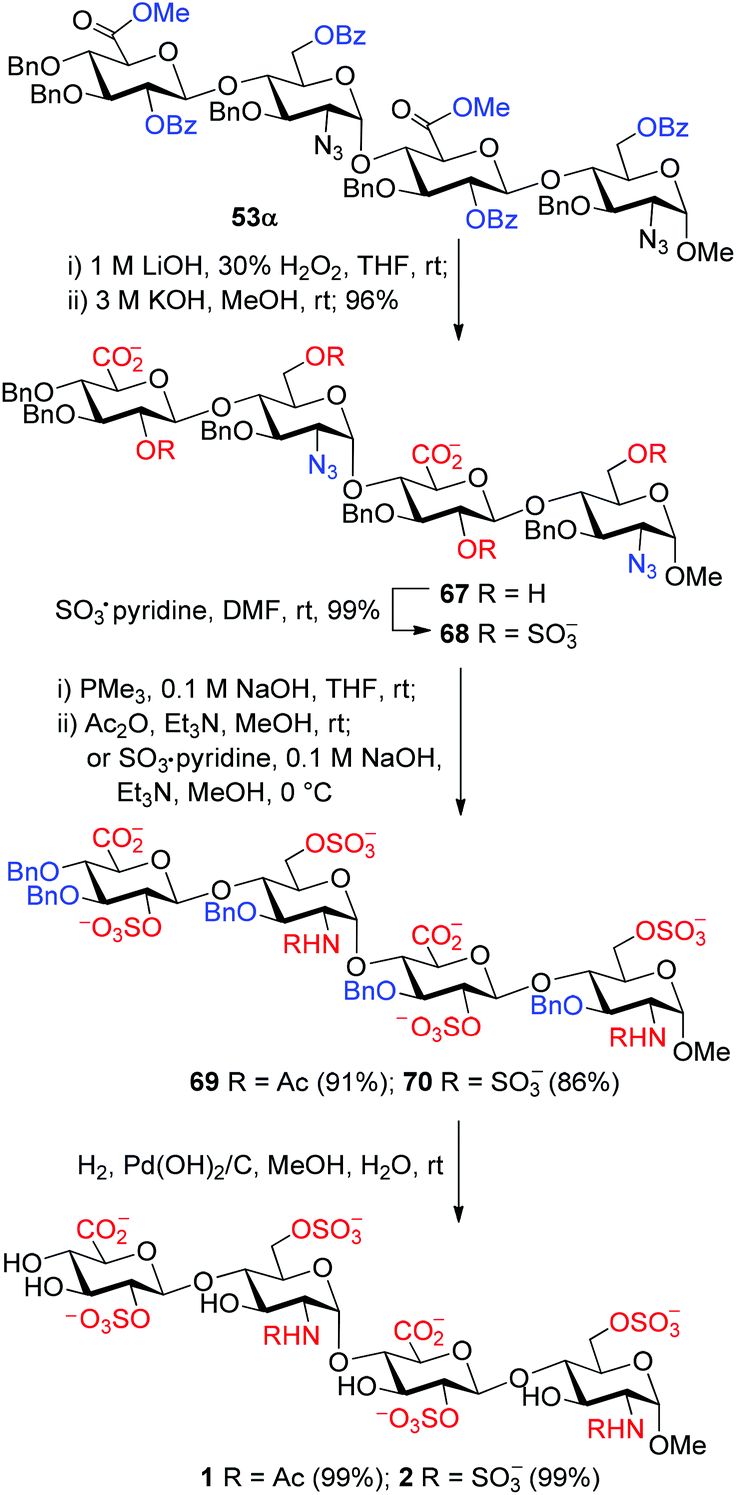 | ||
| Scheme 5 Elaboration of the heparin tetrasaccharides 1 and 2. | ||
 | ||
| Scheme 6 The heparin oligosaccharides (3–20) synthesized from the fully protected precursors. | ||
Conclusion
A library of tri- and tetrasaccharides (1–20) structurally relevant to ΔHexA(2S)–GlcN(NS,6S)–GlcA–GlcN(NS,6S), an optimal substrate of heparanase, has been synthesized efficiently. These HS oligosaccharides are elaborated from the corresponding orthogonally protected tri- and tetrasaccharides (53α–62α) employing robust transformations including saponification, O-sulfation, azide reduction, N-sulfation/N-acetylation, and debenzylation. The advanced oligosaccharide precursors (53α–62α) are synthesized with the [2 + 2] and [1 + 2] glycosylation as the key step in building the difficult α-GlcN–(1→4)-GlcA/IdoA linkages. This particular glycosylation with a variety of the 2-azido-glucopyranosyl donors has been investigated carefully. A standard protocol has been fixed that employs a glycosyl N-phenyltrifluoroacetimidate as a donor, TMSOTf as a catalyst, toluene as a solvent, and −30 °C as the working temperature. With this protocol, a number of coupling partners have been assembled reliably in satisfactory yields and α-selectivity. It is observed that a remote protecting group or a sugar unit in either the donor or the acceptor could affect considerably both the coupling yield and α-selectivity. Nevertheless, the mechanism behind such effects is yet to be elucidated. The availability/accessibility of the HS tri- and tetrasaccharides would enable us to identify an appropriate substrate of heparanase so as to establish an optimal assay for measuring the heparanase activities.Experimental section
General glycosylation procedure for the synthesis of disaccharides (29, 30, 37–39, 47, 50)
The glycosyl imidate donor (1.1 equiv.) and 2-azido-2-deoxy-D-glucopyranoside acceptor (1.0 equiv.) were combined in a flask and dissolved in CH2Cl2 to maintain a concentration of 0.02 M. Freshly activated powdered 4 Å MS (weight equal to the combined weight of the donor and the acceptor) was added. The mixture was stirred for 1 hour at ambient temperature and then cooled to −30 °C. TMSOTf (0.05 equiv.) was added to the mixture, and the stirring was continued until TLC indicated disappearance of the donor (∼30 min). The reaction was quenched by the addition of Et3N. The mixture was filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography using a gradient of petroleum ether–EtOAc to afford the pure product.General procedure for the cleavage of the benzylidene acetal (41–43 and 51)
A solution of the 4′,6′-O-benzylidene disaccharide (0.05 M) in HOAc–H2O (v/v = 4/1) was stirred at 80 °C for 1 h. The mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography using a gradient of petroleum ether–EtOAc to give the pure product.General procedure for the regioselective opening of the benzylidene acetal
BH3·THF (10 equiv.) was added to a solution of the 4′,6′-O-benzylidene disaccharide (29, 30, or 47) in THF at 0 °C. After 15 min, Bu2B·OTf (2 equiv.) was added dropwise and the stirring was continued at 0 °C for 2 h. The reaction was quenched by addition of Et3N and the excess BH3·THF was consumed by slowly adding methanol. The solution was concentrated in vacuo to give the crude product.General procedure for the TEMPO/BAIB-mediated oxidation and methyl ester formation (31, 32, 44–46, 48, 52)
To a vigorously stirring solution of the disaccharide 4′,6′-diol (0.3 M solution) in CH2Cl2–H2O (v/v = 2/1) were added TEMPO (0.1 equiv.) and BAIB (2.5 equiv.). Stirring was continued until TLC indicated complete conversion of the starting material to a spot of lower Rf (∼3 h). The reaction was quenched by the addition of aqueous Na2S2O3 (10 mL). The mixture was acidized with 5% HCl, then extracted with CH2Cl2 three times. The combined organic layers were washed with brine (50 mL), and were then dried over Na2SO4 and filtered. The filtrate was concentrated in vacuo, and the residue was co-evaporated with toluene twice. The resulting oily residue was dissolved in DMF (0.1 M), and then treated with KHCO3 (4 equiv.) and methyl iodide (8 equiv.). The excess methyl iodide was quenched by the addition of AcOH. The mixture was concentrated in vacuo, and then diluted with CH2Cl2. The solution was washed with brine twice and then evaporated. The residue was purified by silica gel column chromatography using a gradient of petroleum ether–EtOAc to yield the methyl ester.General procedure for the cleavage of the silyl ethers
The silyl glycoside (31, 32, or 48) was dissolved in THF (0.1 M) followed by the addition of AcOH (3 equiv.) and TBAF in THF (1 M, 2 equiv.). After stirring for 30 min, the mixture was diluted with CH2Cl2 (50 mL) and washed with saturated aqueous NaHCO3 (50 mL) and brine (50 mL), respectively. The organic phase was dried (Na2SO4) and filtered. The filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography using a gradient of petroleum ether–EtOAc to give the pure lactol.General procedure for the preparation of the glycosyl N-phenyl trifluoroacetimidates (24, 35, 36, 49)
To a solution of the lactol in acetone (2 mL, 0.1 mmol) were added finely powdered anhydrous K2CO3 (2.5 equiv.) and N-phenyl trifluoroacetimidoyl chloride (1.3 equiv.). After stirring at room temperature for 1.5 h, the mixture was filtered. The filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (petroleum ether–EtOAc containing 1% Et3N) to give the pure product.General glycosylation procedure for the preparation of the tri- and tetrasaccharides (53–62)
The glycosyl N-phenyl trifluoroacetimidate donor (1.2 equiv.) and disaccharide acceptor (1.0 equiv.) were combined in a flask, co-evaporated with toluene (3 × 3 mL), and then dissolved in toluene to maintain a concentration of 0.04–0.05 M. Freshly activated powdered 5 Å MS (weight equal to the combined weight of the donor and the acceptor) was added, and the mixture was stirred for 1 h at ambient temperature and then cooled to −30 °C. TMSOTf (0.1 equiv.) was added, and stirring was continued until TLC indicated the disappearance of the donor (∼2 hours). The reaction was quenched by addition of Et3N (0.5 mL). The mixture was filtered. The filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography using a gradient of toluene–EtOAc to give the pure product. The α and β anomers were fully separated.General procedure for the saponification of the methyl esters and benzoyl groups
A premixed solution of 30% solution of H2O2 in water (100 equiv. per CO2Me) and 1 M LiOH (50 equiv. per CO2Me) were added to a solution of the starting material in THF (0.02 M). The mixture was stirred at rt for 24 h. A solution of KOH (3 M) was added until pH ≃ 14. The mixture was left stirring for 24 h at room temperature. The mixture was then brought to pH = 8–8.5 by addition of acidic resin, and then filtered. The filtrate was concentrated in vacuo (bath temperature 20–30 °C). The residue was dissolved in CH2Cl2–MeOH (v/v = 1/1). The resulting solution was layered on the top of a Sephadex LH-20 chromatography column and then eluted with CH2Cl2–MeOH (v/v = 1/1). The appropriate fraction was concentrated in vacuo to provide the pure product.General procedure for the O-sulfation
SO3·pyridine complex (8 equiv. per OH) was added to a solution of the starting material in DMF (1.0 mL, 0.02 mmol). The mixture was stirred at ambient temperature for 4 h until TLC (RP18 silica gel, H2O–CH3OH, v/v = 1/2) indicated completion of the reaction. After addition of CH3OH (0.5 mL), the stirring was continued for 30 min. The mixture was concentrated in vacuo. The residue was vortexed with water and then applied to a small RP-18 silica gel column, which was eluted with a gradient of H2O–CH3OH (v/v from 1/0 to 2/1). The fractions containing the product were concentrated in vacuo. The residue was immediately passed through a column of Dowex 50WX4 Na+ resin using CH3OH as an eluent. The appropriate fraction was concentrated in vacuo to provide the product as sodium salt.General procedure for the reduction of the azide
A solution of Me3P in THF (1 M, 4 equiv. per azide) was added to a solution of the starting material in THF (1.0 mL, 0.02 mmol). An NaOH solution (0.1 M, 5 equiv. per azido) was added, and the mixture was stirred at room temperature for 5 h. The progress of the reaction was monitored by TLC (RP-18 silica gel, H2O–CH3OH, v/v = 1/2). The pH was then adjusted to 7–8 by careful addition of HCl (1.0 M). The mixture was then concentrated in vacuo to give the crude product.General procedure for the N-sulfation
SO3·pyridine (5 equiv. per NH2) was added to the starting material in CH3OH (1 mL, 0.01 mmol) containing triethylamine (0.3 mL) and NaOH (0.1 M, 2 equiv. per NH2) at 0 °C. The progress of the reaction was monitored by TLC (RP-18 silica gel, H2O–CH3OH, v/v = 1/1). Two additional portions of SO3·pyridine were added at 0 °C after 30 min and 1 h, respectively. After stirring for an additional 8 h at 0 °C, the mixture was concentrated in vacuo. The residue was vortexed with water and then applied to a small RP-18 silica gel column, which was eluted with a gradient of H2O–CH3OH (v/v from 1/0 to 1/1). The fractions containing the product were concentrated in vacuo. The residue was immediately passed through a column of Dowex 50WX4 Na+ resin using a mixture of CH3OH and H2O (9/1, v/v) as an eluent. The appropriate fraction was concentrated in vacuo to provide the product.General procedure for the N-acetylation
Acetic anhydride (10 equiv. per NH2) was added to a solution of the starting material in a mixture of anhydrous CH3OH (0.02 mmol) and Et3N (20 equiv. per NH2) at 0 °C. The progress of the reaction was monitored by TLC (RP18 silica gel, H2O–CH3OH, v/v = 1/1). After stirring for 4 h at room temperature, the mixture was concentrated in vacuo. The residue was vortexed with water and applied to a small RP-18 silica gel column, which was eluted with a gradient of H2O–CH3OH (v/v from 1/0 to 1/1). The fractions containing the product were concentrated in vacuo. The residue was immediately passed through a column of Dowex 50WX4 Na+ resin using CH3OH–H2O (v/v = 9/1) as an eluent. The appropriate fraction was concentrated in vacuo to provide the product.General procedure for the global removal of benzyl groups (1–20)
Palladium hydroxide on carbon (Degussa type, 20%, 1.5–2.0 times the weight of the starting material) was added to a solution of the starting material in CH3OH and pH = 7 buffer H2O (1 mL for 10–20 mg, v/v = 1/1). The mixture was placed under an atmosphere of hydrogen for 24 h. The mixture was filtered and concentrated. The residue was diluted with H2O. The solution was layered on the top of a Sephadex G-10 column that was eluted with H2O. The fractions containing the product were concentrated in vacuo. The residue was immediately passed through a column of Dowex 50WX4 Na+ resin using H2O as an eluent. The appropriate fraction was freeze dried to provide the final product as a white solid.Acknowledgements
Financial support from the Ministry of Science and Technology of China (2012ZX09502-002) and the National Natural Science Foundation of China (20932009) is gratefully acknowledged.Notes and references
- (a) I. Capila and R. J. Linhardt, Angew. Chem., Int. Ed., 2002, 41, 390–412 CrossRef CAS; (b) J. D. Esko and S. B. Selleck, Annu. Rev. Biochem., 2002, 71, 435–471 CrossRef CAS PubMed; (c) J. Turnbull, A. Powell and S. Guimond, Trends Cell Biol., 2001, 11, 75–82 CrossRef CAS PubMed; (d) N. S. Gandhi and R. L. Mancera, Chem. Biol. Drug Des., 2008, 72, 455–482 CrossRef CAS PubMed.
- (a) C. A. A. van Boeckel and M. Petitou, Angew. Chem., Int. Ed. Engl., 1993, 32, 1671–1690 CrossRef; (b) M. Petitou, J.-P. Herault, A. Bernat, P.-A. Driguez, P. Duchaussoy, J.-C. Lormeau and J.-M. Herbert, Nature, 1999, 398, 417–422 CrossRef CAS PubMed; (c) B. Casu and U. Lindahl, Adv. Carbohydr. Chem. Biochem., 2001, 57, 159–206 CrossRef CAS PubMed; (d) M. de Kort, R. C. Buijsman and C. A. A. van Boeckel, Drug Discovery Today, 2005, 10, 769–779 CrossRef CAS PubMed; (e) D. Bonnaffé, C. R. Chim., 2011, 14, 59–73 CrossRef.
- (a) U. Hacker, K. Nybakken and N. Perrimon, Nat. Rev. Mol. Cell Biol., 2005, 6, 530–541 CrossRef PubMed; (b) J. M. Whitelock and R. V. Iozzo, Chem. Rev., 2005, 105, 2745–2764 CrossRef CAS PubMed.
- (a) J. T. Gallagher and A. Walker, Biochem. J., 1985, 230, 665–674 CrossRef CAS PubMed; (b) S. E. Stringer and J. T. Gallagher, Int. J. Biochem. Cell Biol., 1997, 29, 709–714 CrossRef CAS PubMed.
- S. Peterson, A. Frick and J. Liu, Nat. Prod. Rep., 2009, 26, 610–627 RSC.
- (a) J. R. Bishop, M. Schuksz and J. D. Esko, Nature, 2007, 446, 1030–1037 CrossRef CAS PubMed; (b) C. R. Parish, Nat. Rev. Immunol., 2006, 6, 633–643 CrossRef CAS PubMed; (c) A. K. Powell, E. A. Yates, D. G. Fernig and J. E. Turnbull, Glycobiology, 2004, 14, 17R–30R CrossRef CAS PubMed; (d) R. J. Linhardt, J. Med. Chem., 2003, 46, 2551–2564 CrossRef CAS PubMed.
- (a) A. Ori, M. C. Wilkinson and D. G. Fernig, Front. Biosci., 2008, 13, 4309–4338 CrossRef CAS PubMed; (b) D. A. Rabenstein, Nat. Prod. Rep., 2002, 19, 312–331 RSC; (c) C. Noti and P. H. Seeberger, Chem. Biol., 2005, 12, 731–756 CrossRef CAS PubMed; (d) U. Lindahl, Glycoconjugate J., 2000, 17, 597–605 CrossRef CAS.
- (a) H. E. Conrad, Heprarin Binding proteins, Academic Press, San Diego, 1998 Search PubMed; (b) H. G. Garg, R. J. Linhardt and C. A. Hales, Chemistry and Biology of Heparin and Heparan Sulfate, Elsevier, Amsterdam, 2005 Search PubMed.
- M. Petitou and C. A. A. van Beockel, Angew. Chem., Int. Ed., 2004, 43, 3118–3133 CrossRef CAS PubMed.
- (a) C. Freeman and C. R. Parish, Biochem. J., 1998, 330, 1341–1350 CrossRef CAS PubMed; (b) I. Vlodavsky and Y. Friedmann, J. Clin. Invest., 2001, 108, 341–347 CrossRef CAS PubMed; (c) C. R. Parish, C. Freeman and M. D. Hulett, Biochim. Biophys. Acta, 2001, 1471, M99–M108 CAS; (d) K. J. Bame, Glycobiology, 2001, 11, 91R–98R CrossRef CAS PubMed; (e) D. S. Pikas, J.-P. Li, I. Vlodavsky and U. Lindahl, J. Biol. Chem., 1998, 273, 18770–18777 CrossRef CAS PubMed; (f) R. D. Sanderson, Y. Yang, T. Kelly, V. MacLeod, Y. Dai and A. Theus, J. Cell. Biochem., 2005, 96, 897–905 CrossRef CAS PubMed.
- M. Elkin, N. Ilan, R. Ishai-Michaeli, Y. Friedmann, O. Papo, I. Pecker and I. Vlodavsky, FAESB J., 2001, 15, 1661–1663 CAS.
- (a) N. Ilan, M. Elkin and I. Vlodavsky, Int. J. Biochem. Cell Biol., 2006, 38, 2018–2039 CrossRef CAS PubMed; (b) M. D. Hulett, C. Freeman, B. J. Hamdorf, R. T. Baker, M. J. Harris and C. R. Parish, Nat. Med., 1999, 5, 803–809 CrossRef CAS PubMed.
- (a) S. Simizu, K. Ishida and H. Osada, Cancer Sci., 2004, 95, 553–558 CrossRef CAS PubMed; (b) H.-Q. Miao, H. Liu, E. Navarro, P. Kussie and Z. Zhu, Curr. Med. Chem., 2006, 13, 2101–2111 CrossRef CAS PubMed.
- (a) Y. Okada, S. Yamada, M. Toyoshima, J. Dong, M. Nakajima and K. Sugahara, J. Biol. Chem., 2002, 277, 42488–42495 CrossRef CAS PubMed; (b) S. B. Peterson and J. Liu, Matrix Biol., 2013, 32, 223–227 CrossRef CAS PubMed.
- (a) S. B. Peterson and J. Liu, J. Biol. Chem., 2010, 285, 14504–14513 CrossRef CAS PubMed; (b) E. Hammond, C. P. Li and V. Ferro, Anal. Biochem., 2010, 396, 112–116 CrossRef CAS PubMed; (c) A. G. Pearson, M. J. Kiefel, V. Ferro and M. von Itzstein, Org. Biomol. Chem., 2011, 9, 4614–4625 RSC; (d) N. Takeda, R. Ikeda-Matsumi, K. Ebara-Nagahara, M. Otaki-Nanjo, K. Taniguchi-Morita, M. Nano and J. Tamura, Carbohydr. Res., 2012, 353, 13–21 CrossRef CAS PubMed.
- (a) P. Sinay, J.-C. Jacquinet, M. Petitou, P. Duchaussoy, I. Lederman, J. Choay and G. Torri, Carbohydr. Res., 1984, 132, C5–C9 CrossRef CAS; (b) C. A. A. van Boeckel, T. Beetz, J. N. Vos, A. J. M. de Jong, S. F. van Aelst, R. H. van den Bosch, J. M. R. Mertens and F. A. van der Vlugt, J. Carbohydr. Chem., 1985, 4, 293–321 CrossRef CAS; (c) M. Petitou, P. Duchaussoy, I. Lederman, J. Choay, P. Sinay, J.-C. Jacquinet and G. Torri, Carbohydr. Res., 1986, 147, 221–236 CrossRef CAS PubMed.
- (a) J.-L. de Paz, J. Angulo, J.-M. Lassaletta, P. M. Nieto, M. Redondo-Horcajo, R. M. Lozano, G. Gimenez-Gallego and M. Martin-Lomas, ChemBioChem, 2001, 2, 673–685 CrossRef CAS PubMed; (b) M. F. Haller and G.-J. Boons, Eur. J. Org. Chem., 2002, 2033–2038 CrossRef CAS; (c) H. A. Orgueira, A. Bartolozzi, P. Schell, R. E. J. N. Litjens, E. R. Palmacci and P. H. Seeberger, Chem. – Eur. J., 2003, 9, 140–169 CrossRef CAS PubMed; (d) L. Poletti and L. Lay, Eur. J. Org. Chem., 2003, 2999–3024 CrossRef CAS; (e) R. Ojeda, J.-L. de Paz and M. Martin-Lomas, Chem. Commun., 2003, 2486–2487 RSC; (f) J.-C. Lee, X.-A. Lu, S. S. Kulkarni, Y.-S. Wen and S.-C. Hung, J. Am. Chem. Soc., 2004, 126, 476–477 CrossRef CAS PubMed; (g) G. J. S. Lohman and P. H. Seeberger, J. Org. Chem., 2004, 69, 4081–4093 CrossRef CAS PubMed; (h) H. N. Yu, J.-i. Furukawa, T. Ikeda and C.-H. Wong, Org. Lett., 2004, 6, 723–4282 CrossRef CAS PubMed; (i) A. Lubineau, H. Lortat-Jacob, O. Gavard, S. Sarrazin and D. Bonnaffe, Chem. – Eur. J., 2004, 10, 4265–4282 CrossRef CAS PubMed; (j) J. D. C. Codee, B. Stubba, M. Schiattarella, H. S. Overkleeft, C. A. A. van Boeckel, J. H. van Boom and G. A. van der Marel, J. Am. Chem. Soc., 2005, 127, 3767–3773 CrossRef CAS PubMed; (k) J. L. de Paz and M. Martin-Lomas, Eur. J. Org. Chem., 2005, 1849–1858 CrossRef CAS.
- (a) C. Noti, J.-L. de Paz, L. Polito and P. H. Seeberger, Chem. – Eur. J., 2006, 12, 8664–8686 CrossRef CAS PubMed; (b) L.-D. Lu, C.-R. Shie, S. S. Kulkarni, G.-R. Pan, X.-A. Lu and S.-C. Hung, Org. Lett., 2006, 8, 5995–5998 CrossRef CAS PubMed; (c) J.-L. de Paz, C. Noti and P. H. Seeberger, J. Am. Chem. Soc., 2006, 128, 2766–2767 CrossRef CAS PubMed; (d) A. Adibekian, P. Bindschädler, M. S. M. Timmer, C. Noti, N. Schützenmeister and P. H. Seeberger, Chem. – Eur. J., 2007, 13, 4510–4522 CrossRef CAS PubMed; (e) T. Polat and C.-H. Wong, J. Am. Chem. Soc., 2007, 129, 12795–12800 CrossRef CAS PubMed; (f) J. Chen, Y. Zhou, C. Chen, W. Xu and B. Yu, Carbohydr. Res., 2008, 343, 2853–2862 CrossRef CAS PubMed; (g) F. Baleux, L. Loureiro-Morais, Y. Hersant, P. Clayette, F. Arenzana-Seisdedos, D. Bonnaffe and H. Lortat-Jacob, Nat. Chem. Biol., 2009, 5, 743–748 CrossRef CAS PubMed; (h) S. Arungundram, K. Al-Mafraji, J. Asong, F. E. Leach, I. J. Amster, A. Venot, J. E. Turnbull and G.-J. Boons, J. Am. Chem. Soc., 2009, 131, 17394–17405 CrossRef CAS PubMed; (i) Z. Wang, Y. Xu, B. Yang, G. Tiruchinapally, B. Sun, R. Liu, S. Dulaney, J. Liu and X. Huang, Chem. – Eur. J., 2010, 16, 8365–8375 CrossRef CAS PubMed; (j) G. Tiruchinapally, Z. Yin, M. El-Dakdouki, Z. Wang and X. Huang, Chem. – Eur. J., 2011, 17, 10106–10112 CrossRef CAS PubMed; (k) Y.-P. Hu, S.-Y. Lin, C.-Y. Huang, M. M. L. Zulueta, J.-Y. Liu, W. Chang and S.-C. Hung, Nat. Chem., 2011, 3, 557–563 CrossRef CAS PubMed; (l) M. M. L. Zulueta, S.-Y. Lin, Y.-T. Lin, C.-J. Huang, C.-C. Wang, C.-C. Ku, Z. Shi, C.-L. Chyan, D. Irene, L.-H. Lim, T.-I. Tsai, Y.-P. Hu, S. D. Arco, C.-H. Wong and S.-C. Hung, J. Am. Chem. Soc., 2012, 134, 8988–8995 CrossRef CAS PubMed; (m) Y.-P. Hu, Y.-Q. Zhong, Z.-G. Chen, C. Y. Chen, Z. Shi, M. M. L. Zulueta, C.-C. Ku, P.-Y. Lee, C.-C. Wang and S.-C. Hung, J. Am. Chem. Soc., 2012, 134, 20722–20727 CrossRef CAS PubMed; (n) S.-C. Hung, X.-A. Lu, J.-C. Lee, M. D.-T. Chang, S.-l. Fang, T.-c. Fan, M. M. L. Zulueta and Y.-Q. Zhong, Org. Biomol. Chem., 2012, 10, 760–772 RSC; (o) S. U. Hansen, G. J. Miller, M. Barath, K. R. Broberg, E. Avizienyte, M. Helliwell, J. Raftery, G. C. Jayson and J. M. Gardiner, J. Org. Chem., 2012, 77, 7823–7843 CrossRef CAS PubMed; (p) S. U. Hansen, G. J. Miller, G. C. Jayson and J. M. Gardiner, Org. Lett., 2013, 15, 88–91 CrossRef CAS PubMed; (q) N. Guedes, P. Czechura, B. Echeverria, A. Ruiz, O. Michelena, M. Martin-Lomas and N.-C. Reichardt, J. Org. Chem., 2013, 78, 6911–6934 CrossRef CAS PubMed; (r) C. Zong, A. Venot, O. Dhamale and G.-J. Boons, Org. Lett., 2013, 15, 342–345 CrossRef CAS PubMed.
- (a) Y. Xu, S. Masuko, M. Takieddin, H. Xu, R. Liu, J. Jing, S. A. Mousa, R. J. Linhardt and J. Liu, Science, 2011, 334, 498–501 CrossRef CAS PubMed; (b) R. Liu, Y. Xu, M. Chen, M. Weiwer, X. Zhou, A. S. Bridges, P. L. DeAngelis, Q. Zhang, R. J. Linhardt and J. Liu, J. Biol. Chem., 2010, 285, 34240–34249 CrossRef CAS PubMed.
- (a) A. F. G. Bongat and A. V. Demchenko, Carbohydr. Res., 2007, 342, 374–406 CrossRef CAS PubMed; (b) R. Enugala, L. C. R. Carvalho, M. J. Dias Pires and M. M. B. Marques, Chem. – Asian J., 2012, 7, 2482–2501 CrossRef CAS PubMed.
- (a) H. Paulsen, C. Kolar and W. Stenzel, Angew. Chem., Int. Ed. Engl., 1976, 15, 440–441 CrossRef CAS PubMed; (b) H. A. Orgueira, A. Bartolozzi, P. Schell and P. H. Seeberger, Angew. Chem., Int. Ed., 2002, 41, 2128–2131 CrossRef CAS.
- (a) K. Benakli, C. Zha and R. J. Kerns, J. Am. Chem. Soc., 2001, 123, 9461–9462 CrossRef CAS PubMed; (b) M. Boysen, E. Gemma, M. Lahmann and S. Oscarson, Chem. Commun., 2005, 3044–3046 RSC; (c) Y. Geng, L.-H. Zhang and X.-S. Ye, Chem. Commun., 2008, 597–599 RSC; (d) S. Manabe, K. Ishii and Y. Ito, J. Am. Chem. Soc., 2006, 128, 10666–10667 CrossRef CAS PubMed.
- (a) E. A. Mensah and H. M. Nguyen, J. Am. Chem. Soc., 2009, 131, 8778–8780 CrossRef CAS PubMed; (b) E. A. Mensah, F. Yu and H. M. Nguyen, J. Am. Chem. Soc., 2010, 132, 14288–14302 CrossRef CAS PubMed.
- (a) M. B. Cid, F. Alfonso and M. Martin-Lomas, Chem. – Eur. J., 2005, 11, 928–938 CrossRef CAS PubMed; (b) J. Banoub, P. Boullanger and D. Lafont, Chem. Rev., 1992, 92, 1167–1195 CrossRef CAS; (c) B. Fraser-Reid, K. N. Jayaprakash, J. C. Lopez, A. M. Gomez and C. Uriel, Protecting Groups in Carbohydrate Chemistry Profoundly Influence All Selectivities in Glycosyl Couplings, in Frontiers in Modern Carbohydrate Chemistry. ACS Symposium Series 960, American Chemical Society, Washington, DC, 2007, pp. 91–117 Search PubMed.
- (a) K. Lidholt and U. Lindahl, Biochem. J., 1992, 287, 21–29 CrossRef CAS PubMed; (b) U. Lindahl, M. Kusche-Gullberg and L. Kjellen, J. Biol. Chem., 1998, 273, 24979–24982 CrossRef CAS PubMed; (c) J. D. Esko and U. Lindahl, J. Clin. Invest., 2001, 108, 169–173 CrossRef CAS PubMed; (d) T. B. Feyerabend, J.-P. Li, U. Lindahl and H.-R. Rodewald, Nat. Chem. Biol., 2006, 2, 195–196 CrossRef CAS PubMed.
- M. Bernfield, M. Gotte, P. W. Park, O. Reizes, M. L. Fitzgerald, J. Lincecum and M. Zako, Annu. Rev. Biochem., 1999, 68, 729–777 CrossRef CAS PubMed.
- (a) J. H. van Boom and P. M. J. Burgers, Tetrahedron Lett., 1976, 4875–4878 CrossRef CAS; (b) T. Zhu and G.-J. Boons, Tetrahedron: Asymmetry, 2000, 11, 199–205 CrossRef CAS.
- (a) G.-J. Boons, Contemp. Org. Synth., 1996, 3, 173–200 RSC; (b) A. V. Demchenko, Synlett, 2003, 1225–1240 CrossRef CAS.
- (a) A. De Mico, R. Margarita, L. Parlanti, A. Vescovi and G. Piancatelli, J. Org. Chem., 1997, 62, 6974–6977 CrossRef CAS; (b) J. B. Epp and T. S. Widlanski, J. Org. Chem., 1999, 64, 293–295 CrossRef CAS PubMed; (c) L. J. van den Bos, J. D. C. Codee, J. C. van der Toorn, T. J. Boltje, J. H. van Boom, H. S. Overkleeft and G. A. van der Marel, Org. Lett., 2004, 6, 2165–2168 CrossRef CAS PubMed.
- (a) Y. Zhou, F. Lin, J. Chen and B. Yu, Carbohydr. Res., 2006, 341, 1619–1629 CrossRef CAS PubMed; (b) C. Chen and B. Yu, Bioorg. Med. Chem. Lett., 2009, 19, 3875–3879 CrossRef CAS PubMed.
- See ESI† for details.
- R. R. Schmidt, Angew. Chem., Int. Ed. Engl., 1986, 25, 212–235 CrossRef.
- L. Jiang and T. Chan, Tetrahedron Lett., 1998, 39, 355–358 CrossRef CAS.
- (a) C. Wang, S. Luo, C. Shie and S.-C. Hung, Org. Lett., 2002, 4, 847–849 CrossRef CAS PubMed; (b) J. Hernandez-Torres, J. Achkar and A. Wei, J. Org. Chem., 2004, 69, 7206–7211 CrossRef CAS PubMed; (c) C. Shie, Z. Tzeng, S. Kulkarni, B. Uang, C. Hsu and S.-C. Hung, Angew. Chem., Int. Ed., 2005, 44, 1665–1669 CrossRef CAS PubMed.
- (a) Y. Li, Y. Yang and B. Yu, Tetrahedron Lett., 2008, 49, 3604–3608 CrossRef CAS; (b) Y. Yang, Y. Li and B. Yu, J. Am. Chem. Soc., 2009, 131, 12076–12077 CrossRef CAS PubMed; (c) Y. Li, X. Yang, Y. Liu, C. Zhu, Y. Yang and B. Yu, Chem. – Eur. J., 2010, 16, 1871–1882 CrossRef CAS PubMed; (d) Y. Zhu and B. Yu, Angew. Chem., Int. Ed., 2011, 50, 8329–8332 CrossRef CAS PubMed; (e) Y. Tang, J. Li, Y. Zhu, Y. Li and B. Yu, J. Am. Chem. Soc., 2013, 135, 18396–18405 CrossRef CAS PubMed.
- (a) B. Yu and H. Tao, Tetrahedron Lett., 2001, 42, 2405–2407 CrossRef CAS; (b) B. Yu and H. Tao, J. Org. Chem., 2002, 67, 9099–9102 CrossRef CAS PubMed; (c) B. Yu and J. Sun, Chem. Commun., 2010, 46, 4668–4679 RSC.
- (a) M. Petitou, P. Duchaussoy, P. A. Driguez, G. Jaurand, J.-P. He'rault, J.-C. Lormeau, C. A. A. van Boeckel and J. M. Herbert, Angew. Chem., Int. Ed., 1998, 37, 3009–3014 CrossRef CAS; (b) P. Duchaussoy, G. Jaurand, P. A. Driguez, I. Lederman, F. Gourvenec, J.-M. Strassel, P. Sizun, M. Petitou and J. M. Herbert, Carbohydr. Res., 1999, 317, 63–84 CrossRef CAS PubMed.
- R. Lucas, D. Hamza, A. Lubineau and D. Bonnaffe, Eur. J. Org. Chem., 2004, 2107–2117 CrossRef CAS.
- J. Park, S. Kawatkar, J. Kim and G.-J. Boons, Org. Lett., 2007, 9, 1959–1962 CrossRef CAS PubMed.
- N. M. Spijker and C. A. A. Van Boeckel, Angew. Chem., Int. Ed. Engl., 1991, 30, 180–183 CrossRef.
- (a) Z. Zhang, I. R. Ollman, X.-S. Ye, R. Wischnat, T. Baasov and C.-H. Wong, J. Am. Chem. Soc., 1999, 121, 734–753 CrossRef CAS; (b) N. Teumelsan and X. Huang, J. Org. Chem., 2007, 72, 8976–8979 CrossRef CAS PubMed.
- (a) H. Lucas, J. E. M. Basten, T. G. van Dinther, D. G. Meuleman, S. F. van Aelst and C. A. A. van Boeckel, Tetrahedron, 1990, 46, 8207–8228 CrossRef CAS; (b) L. Rochepeau-Jobron and J. C. Jacquinet, Carbohydr. Res., 1998, 305, 181–191 CrossRef.
- (a) M. Brewer and D. H. Rich, Org. Lett., 2001, 3, 945–948 CrossRef CAS PubMed; (b) P. B. Alper, M. Hendrix, P. Sears and C.-H. Wong, J. Am. Chem. Soc., 1998, 120, 1965–1978 CrossRef CAS.
- K. Horita, T. Yoshioka, T. Tanaka, Y. Oikawa and O. Yonemitsu, Tetrahedron, 1986, 42, 3021–3028 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00039k |
| This journal is © the Partner Organisations 2014 |