A non-diazo strategy to cyclopropanation via oxidatively generated gold carbene: The benefit of a conformationally rigid P,N-bidentate ligand†
Kegong
Ji
and
Liming
Zhang
*
Department of Chemistry and Biochemistry, University of California, Santa Barbara, California, USA. E-mail: zhang@chem.ucsb.edu; Web: http://www.chem.ucsb.edu/~zhang/index.html Fax: (+1) 805-893-4120
First published on 13th February 2014
Abstract
What a trade: diazo ketones for alkynes? By utilizing a conformationally rigid P,N-bidentate ligand, highly efficient intramolecular cyclopropanation reactions using flexible and electronically neutral terminal alkyne substrates are realized via a sterically shielded tri-coordinated gold carbene intermediate. Bicyclic/tricyclic functionalized cyclopropyl ketones are formed in mostly good yields in three steps from readily available enones/enals, which compares favorably with related strategies based on the diazo approach in terms of step economy and operational safety. With this chemistry, we have expanded alkynes as surrogates of α-diazo ketones to cyclopropanation reactions.
One of the salient and powerful transformations of α-oxo metal carbene/carbenoids is cyclopropanation of alkenes, especially those without electronic bias. The use of late transition metals such as Rh in this area are particularly prolific.1 Among the various approaches to generating these reactive metal carbene intermediates, decomposition of α-diazo carbonyl compounds is the most reliable, straightforward and utilized one.1a Owing to the energetic diazo moiety, however, these precursors are considered hazardous and potentially explosive; moreover, their preparations also require energetic reagents and frequently multiple steps.
A few years ago we initiated a program of generating α-oxo gold carbene intermediates via gold-catalyzed intermolecular alkyne oxidation2 and exploiting their reactivities (Scheme 1A). This approach circumvents the use of hazardous α-diazo carbonyl precursors and thereby constitutes a highly valuable non-diazo approach in α-oxo metal carbene chemistry. Indeed, an array of useful synthetic methods have been developed by us3 and other researchers,4 which firmly establishes that a C–C triple bond is an effective surrogate of an α-diazo carbonyl in the realm of gold catalysis. However, the putative α-oxo gold carbene moiety behaves mostly as a highly electrophilic species3b–d and, in contrast to its versatile Rh counterpart, seldom undergoes valuable cyclopropanation or C–H insertion5 reactions, which likely tempers the enthusiasm for this gold oxidative chemistry.
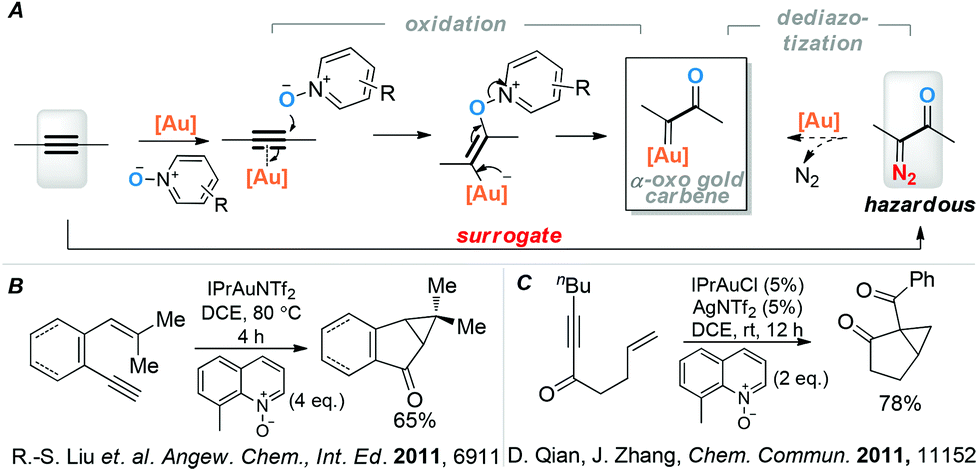 | ||
| Scheme 1 (A) Alkyne as surrogate of α-diazo carbonyl in gold carbene chemistry. (B) Liu's example of cyclopropanation by α-oxo gold carbene intermediates. (C) Zhang's gold-catalyzed oxidative cyclopropanation. | ||
In the case of intramolecular cyclopropanation, there are only two reported cases. Liu and co-workers4c for the first time realized cyclopropanations with dienyne systems (e.g., Scheme 1B), which can be considered special due to their nature of extensive conjugation and conformation rigidity. Later, Qian and Zhang4d reported a gold catalyzed oxidative cyclopropanation of more flexible enyne systems (e.g., Scheme 1C), but the substrates are limited to electron-deficient C–C triple alkynes and an alternative mechanism is not ruled out.
We have recently shown that the high electrophilicity of the α-oxo gold carbene can be attenuated by using P,N-bidentate ligands via the formation of tri-coordinated α-oxo gold carbene intermediates.3e,f With a keen intention to expand alkynes as surrogates of α-diazo carbonyls to highly valuable transformations beyond trapping by NuH, and considering the rather limited success with the cyclopropanation reaction, we wondered whether such less electrophilic gold carbenes could behave better in cyclopropanating tethered alkenes with more flexible and/or general systems. Herein, we report our results and the realization of a highly efficient synthesis of bi-/tricyclic strained ketones in three steps from enones/enals.
At the outset, the TBS-protected 1,5-enyne 1a was chosen as the substrate for the following considerations: (a) it was easily prepared in two steps from cyclohex-2-en-1-one in a 75% combined yield; (b) we suspect that the classical enyne cycloisomerization6 would be minimized due to structural constraints;7 (c) the tricyclic ketone product, i.e., 2a, with its strained and functional structure, appears to be synthetically useful. We initially examined its ability to undergo cycloisomerization in the presence of various gold catalysts. No expected tricyclic alkene 2a′ was detected. With 8-methylquinoline N-oxide3a as the oxidant, the reaction discovery and optimization are detailed in Table 1. Consistent with our past experience, little 2a was detected when typical gold catalysts including Ph3PAuCl/NaBArF4 (entry 1), IPrAuCl/NaBArF4 (entry 2) and [(2,4-tBu2PhO)3P]AuCl/NaBArF4 (entry 3) were used; moreover, the substrate was mostly untouched. With bulky BrettPhos or Me4tBuXPhos as the metal ligand, 2a was indeed formed albeit in <40% yield (entries 4 and 5). These results indicate that steric congestion around the metal center promotes this intramolecular reaction. We then turned to Mor-DalPhos, a P,N-bidentate ligand.8 As expected, the gold catalysis was indeed significantly more efficient than in the case of Me4tBuXPhos although it is smaller (see the %Vbur values9 in Table 1 footnote), consistent with our previously observed beneficial role of a pendant coordinating nitrogen, presumably by facilitating the formation of tri-coordinated gold center.3e,f The smaller Me-DalPhos was expectedly less efficient (entry 8), which clearly points the direction of further improving this reaction. In our previous work on trapping α-oxo gold carbene with carboxylic acids,3f we have demonstrated the benefit of rigidifying the conformation of the pendant piperidine ring. Pleasingly, the same trend was observed in this cyclopropanation chemistry (comparing entries 7–9), and the ligand L2 with a pendant 3,5-cis-dimethylpiperidine ring permitted a highly efficient reaction (entry 9). While the %VBur values are essentially the same for Mor-DalPhos and L2, the latter ligand, owing to its conformation rigidity, offers better shielding of the putative tri-coordinated gold carbene center, thereby behaving as a bulkier ligand. These results reflect the limit of using %VBurto quantify ligand sterics and the importance of considering conformation in ligand design. On the other hand, the diastereomeric ligand L3 with an intruding axial methyl group led to a much lower yield (entry 10). It is important to point out that with Ph2SO as the oxidant10 no 2a was formed (entry 11), again arguing against the alternative mechanism of sequential cycloisomerization and oxidation. The bulky TBS group is important for the reaction efficiency as a much lower yield was observed when replaced by a smaller TES (entry 12). This can be understood by its role of conformation control and its shielding of the oxygen atom from being approached by the electrophilic gold carbene center.2
|
|
|||
|---|---|---|---|
| Entry | R | Catalyst | Yieldb |
a The reaction was run with everything in a vial capped with a septum. Initially, [1a] = 0.1 M.
b Of the gold catalysis step measured by 1H NMR analysis using diethyl phthalate as the internal standard.
c >80% of SM was recovered.
d 91% isolated yield.
e Ph2SO was used as the oxidant, and >80% of SM was recovered. TES = triethylsilyl; TBS = tert-butyldimethylsilyl. 
|
|||
| 1 | TBS | PPh3AuCl (5%)/NaBArF4 (7.5%) | <2c |
| 2 | TBS | IPrAuCl (5%)/NaBArF4 (7.5%) | <2c |
| 3 | TBS | [(2,4-tBu2PhO)3P]AuCl (5%)/NaBArF4 (7.5%) | <2c |
| 4 | TBS | BrettPhosAuCl (5%)/NaBArF4 (7.5%) | 30% |
| 5 | TBS | Me4tBuXPhosAuCl (5%)/NaBArF4 (7.5%) | 34% |
| 6 | TBS | Me-DalPhosAuCl (5%)/NaBArF4 (7.5%) | 30% |
| 7 | TBS | Mor-DalPhosAuCl (5%)/NaBArF4 (7.5%) | 45% |
| 8 | TBS | L1AuCl (5%)/NaBArF4 (7.5%) | 70% |
| 9 | TBS | L2 AuCl (5%)/NaBArF4 (7.5%) | 92% |
| 10 | TBS | L3AuCl (5%)/NaBArF4 (7.5%) | 24% |
| 11 | TBS | L2AuCl (5%)/NaBArF4 (7.5%) | 0%e |
| 12 | TES | L2AuCl (5%)/NaBArF4 (7.5%) | 58% |
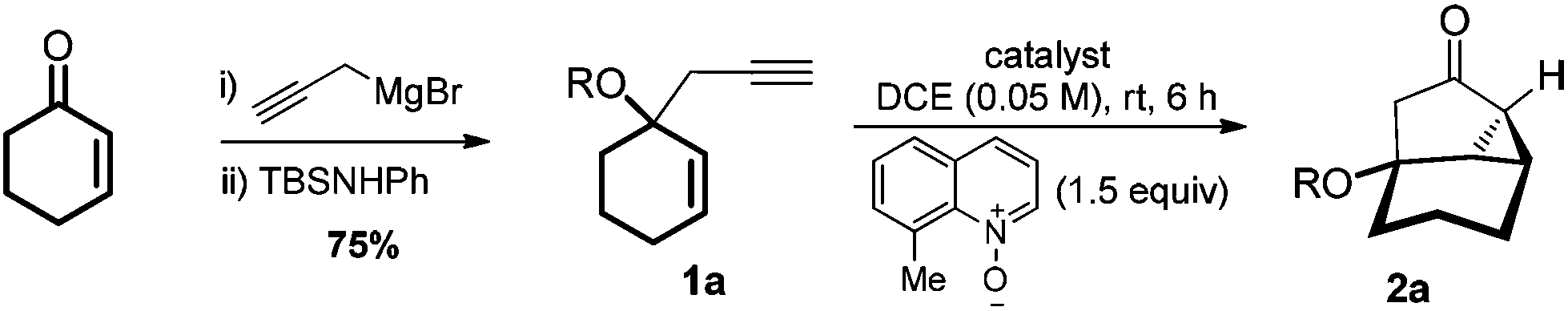
From a synthetic point of view, this gold catalysis permits a three-step sequence to access the functionalized and strained tricycle 2a in a 69% overall yield. In comparison, a sequence to a related product possessing a methyl in place of the OTBS group requires six steps, let alone the need of using highly hazardous diazomethane.11
With the optimized reaction conditions revealed in Table 1, entry 9, the reaction scope was first examined with substrates containing different substituents on the cyclohexene ring. As shown in Table 2, entries 1–4, methyl groups at various positions were allowed, and the corresponding tricyclic ketone products were formed in good to excellent yields. In entry 4 where the substrate was prepared as an isolable diastereoisomer from 6-methylcyclohexan-2-en-1-one in two steps, the hindered tertiary alcohol intermediate could only be protected by a smaller TES group owing to the neighboring Me group, but the reaction remained highly efficient. A vinyl group (entry 5) or phenyl group (entry 6) conjugating to the ring double bond was readily allowed, and the structure of the latter case (i.e., 2g) was confirmed by single crystal X-ray diffraction (Fig. 1).12 The substrate derived from carvone also worked, albeit a relatively low yield (entry 7). This chemistry was readily extended to substrates derived from cyclopent-2-en-1-one (entry 8) and cyclohept-2-en-1-one (entry 9), and the products with slightly different skeletons were formed in good to excellent yields. The relative stereochemistries of all the products are confirmed by extensive 2D NMR studies.
 | ||
| Fig. 1 ORTEP drawing of 2g with the thermal ellipsoids shown at 50% probability. | ||


The reaction also proceeded smoothly with more flexible 3-silyloxy-1,5-enynes, which were again easily prepared from acyclic enones and enals in two steps, and the bicyclo[3.1.0]hexan-2-ones 4 were formed in mostly excellent yields (Table 3). When enones were the ultimate starting material in the three-step sequence, TBS (t-butyldimethylsilyl) was used to protect the HO group; on the other hand, in the case of enals, the bigger TIPS (triisopropylsilyl) led to better yields in the gold catalysis step and hence was preferred. The reaction sequence tolerated various substituted C–C double bonds, including conjugated ones such as β-styryl (entries 1–3) and phenylbuta-1,3-dien-1-yl (entry 9), trisubstituted ones (entries 5, 6 and 7), and disubstituted ones with sensitive functional groups (entry 10). In the cases of substrates prepared from (Z)-β-styryl ketones (entries 1 and 2), good diastereoselectivities (dr ∼ 11) were observed, and extensive NMR studies established that the major diastereomers were the ones with the TBSO group on the concave face. For the other cases, the diastereomeric ratios (dr) were low, and the structures of the major isomers are assigned based on NOESY and are shown in Table 3.


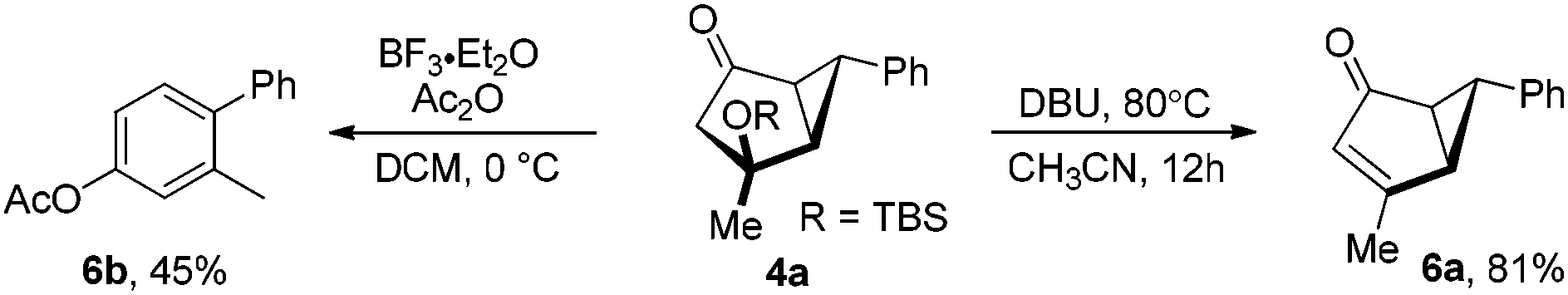
This 3-step, efficient access to the functionalized yet strained cyclic systems opens new and unconventional approaches to synthetically valuable skeletons. For example, the treatment of 2a with BF3·Et2O and Ac2O at 0 °C13 resulted in the formation of the bicyclic enol acetate 5a with completely defined relative stereochemistry at both its ring juncture and the neighboring acetoxy-substituted carbon center, indicating concerted opening of the cyclopropane ring and backside attack by AcO− (eqn (1)). Under the same conditions, 2b, differing from 2a by an additional methyl group on the cyclopropane ring, was transformed into the bicyclic enone 5b in a fairly good yield. Notably, the methyl group is positioned exclusively trans to the TBSO group, opposite to the AcO group in 5a. Its formation can be rationalized by concerted opening of the cyclopropane ring and 1,2-migration of the ring juncture hydride. Eqn (2) exemplifies two transformations of the bicycle[3.1.0]hexan-2-one 4a prepared from acyclic β-styryl methyl ketone. When it was subjected to basic conditions, elimination of a silanol gives the bicyclic cyclopentenone 6a in an 81% yield. Upon subjecting it to the same acidic conditions as in eqn (1), a substituted biphenyl (i.e., 6b) was isolated. Notably, due to the removal of stereogenic centers in both of these reactions, the issues of low diastereoselectivities with linear enone/enals could have no consequence.
 | (1) |
 | (2) |
In conclusion, cyclopropanation of alkenes is a versatile and classic reaction of α-oxo carbenes of late transition metals, but it has only been realized in the case of gold by those generated via oxidation of special types of alkynes. By utilizing a conformationally rigid P,N-bidentate ligand, we achieved highly efficient intramolecular cyclopropanation reactions with flexible and electronically neutral terminal alkynes. Bicyclic/tricyclic functionalized cyclopropyl ketones are formed in mostly good yields in three steps from readily available enones/enals, which compares favorably with related strategies based on the diazo approach in terms of step economy and operational safety. With this chemistry, we have expanded alkynes as surrogates of α-diazo ketones to cyclopropanation reactions. The optimal ligand facilitates the reaction by attenuating the reactivity of the gold carbene via the formation of tri-coordinated gold center and by offering better steric shielding due to conformational rigidity. Further application of this ligand in gold carbene chemistry will be reported in due course.
Experimental section
General procedure for gold-catalyzed oxidative cyclopropanation
To a 3 dram vial containing 6 mL of DCE were added sequentially a 3-silyloxy-1,5-enyne 1 or 3 (0.3 mmol), 8-methylquinoline N-oxide (71.6 mg, 0.45 mmol), L2AuCl (10.8 mg, 0.015 mmol) and NaBArF4 (19.8 mg, 0.0225 mmol). The resulting mixture was stirred at room temperature and the reaction was monitored by TLC. Upon completion, the reaction mixture was concentrated under vacuum. The residue was purified by chromatography on silica gel (eluent: hexanes–ethyl acetate) to afford the desired product 2 or 4.The authors thank NIGMS (R01 GM084254) for generous financial support.
References
- (a) M. P. Doyle, M. A. McKervey and T. Ye, Modern catalytic methods for organic synthesis with diazo compounds: from cyclopropanes to ylides, Wiley, New York, 1998 Search PubMed; (b) D. F. Taber, in Carbon-Carbon σ-Bond Formation, ed. G. Pattenden, Pergamon Press, Oxford, England, New York, 1991, vol. 3, pp. 1045–1062 Search PubMed.
- L. Ye, L. Cui, G. Zhang and L. Zhang, J. Am. Chem. Soc., 2010, 132, 3258–3259 CrossRef CAS PubMed.
- (a) B. Lu, C. Li and L. Zhang, J. Am. Chem. Soc., 2010, 132, 14070–14072 CrossRef CAS PubMed; (b) L. Ye, W. He and L. Zhang, J. Am. Chem. Soc., 2010, 132, 8550–8551 CrossRef CAS PubMed; (c) L. Ye, W. He and L. Zhang, Angew. Chem., Int. Ed., 2011, 50, 3236–3239 CrossRef CAS PubMed; (d) W. He, C. Li and L. Zhang, J. Am. Chem. Soc., 2011, 8482–8485 CrossRef CAS PubMed; (e) Y. Luo, K. Ji, Y. Li and L. Zhang, J. Am. Chem. Soc., 2012, 134, 17412–17415 CrossRef CAS PubMed; (f) K. Ji, Y. Zhao and L. Zhang, Angew. Chem., Int. Ed., 2013, 52, 6508–6512 CrossRef CAS PubMed; (g) Y. Wang, K. Ji, S. Lan and L. Zhang, Angew. Chem., Int. Ed., 2012, 51, 1915–1918 CrossRef CAS PubMed.
- (a) C.-F. Xu, M. Xu, Y.-X. Jia and C.-Y. Li, Org. Lett., 2011, 13, 1556–1559 CrossRef CAS PubMed; (b) A. Mukherjee, R. B. Dateer, R. Chaudhuri, S. Bhunia, S. N. Karad and R.-S. Liu, J. Am. Chem. Soc., 2011, 133, 15372–15375 CrossRef CAS PubMed; (c) D. Vasu, H.-H. Hung, S. Bhunia, S. A. Gawade, A. Das and R.-S. Liu, Angew. Chem., Int. Ed., 2011, 6911–6914 CrossRef CAS PubMed; (d) D. Qian and J. Zhang, Chem. Commun., 2011, 47, 11152–11154 RSC; (e) P. W. Davies, A. Cremonesi and N. Martin, Chem. Commun., 2011, 47, 379–381 RSC; (f) R. B. Dateer, K. Pati and R.-S. Liu, Chem. Commun., 2012, 48, 7200–7202 RSC; (g) W. Yuan, X. Dong, Y. Wei and M. Shi, Chem.–Eur. J., 2012, 18, 10501–10505 CrossRef CAS PubMed; (h) G. Henrion, T. E. J. Chavas, X. Le Goff and F. Gagosz, Angew. Chem., Int. Ed., 2013, 52, 6277–6282 CrossRef CAS PubMed.
- S. Bhunia, S. Ghorpade, D. B. Huple and R.-S. Liu, Angew. Chem., Int. Ed., 2012, 2939–2942 CrossRef CAS PubMed.
- E. s. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef PubMed.
- For exceptions with related but differently substituted enynes, see: (a) V. Mamane, T. Gress, H. Krause and A. Fürstner, J. Am. Chem. Soc., 2004, 126, 8654–8655 CrossRef CAS PubMed; (b) V. López-Carrillo, N. Huguet, Á. Mosquera and A. M. Echavarren, Chem.–Eur. J., 2011, 10972–10978 CrossRef.
- R. J. Lundgren, B. D. Peters, P. G. Alsabeh and M. Stradiotto, Angew. Chem., Int. Ed., 2010, 49, 4071–4074 CrossRef CAS PubMed.
- A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 2009, 1759–1766 CrossRef.
- C. A. Witham, P. Mauleon, N. D. Shapiro, B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 5838–5839 CrossRef CAS PubMed.
- A. Srikrishna, S. Nagaraju and G. V. R. Sharma, J. Chem. Soc., Chem. Commun., 1993, 285–288 RSC.
- The cif file has been deposited at the Cambridge Crystallographic Data Centre (CCDC 957441).
- J. H. Rigby and C. Senanayake, J. Org. Chem., 1988, 53, 440–443 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 957441. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3qo00080j |
| This journal is © the Partner Organisations 2014 |