Solvent-driven selective π-cation templating in dynamic assembly of interlocked molecules†
Andrew B.
Pun
a,
Kevin J.
Gagnon
b,
Liana M.
Klivansky
a,
Simon J.
Teat
b,
Zhan-Ting
Li
*c and
Yi
Liu
*a
aThe Molecular Foundry, Lawrence Berkeley National Laboratory, One Cyclotron Rd, MS 67R6110, Berkeley, CA 94720, USA. E-mail: yliu@lbl.gov; ztli@fudan.edu.cn
bAdvanced Light Source, Lawrence Berkeley National Laboratory, One Cyclotron Rd, MS 67R6110, Berkeley, CA 94720, USA
cDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai 200433, China
First published on 31st January 2014
Abstract
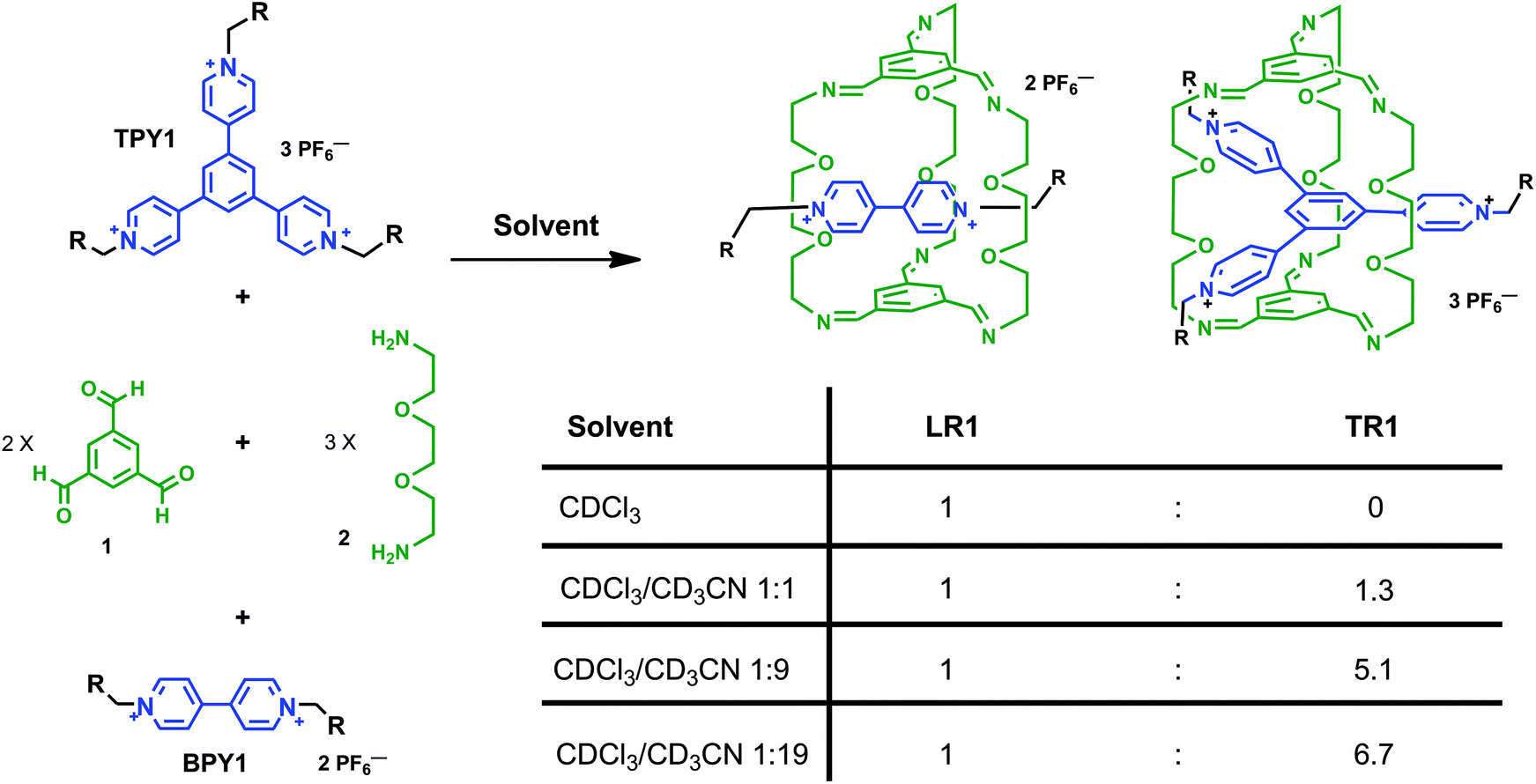
Both bispyridinium (BPY) and trispyridinium (TPY) have been used to template the formation of linear or triply threaded [2]rotaxanes through imine-based dynamic clipping reactions. In this paper, we report contrasting solvent dependence between these two templated clipping reactions when two different solvents, namely CDCl3 and CD3CN, are used. The solvent dependence is elucidated based on 1H NMR studies, and structural features are revealed by single crystal X-ray analyses of the respective linear and triply threaded interlocked molecules. We have shown that although both clipping reactions are affected by hydrogen-bonding and aromatic–aromatic interactions in general, the nature of the aromatic–aromatic interactions is quite different, which is responsible for the different solvent response. The BPY-based clipping reaction is driven by electrostatic interactions between aromatic surfaces, while the TPY-based reaction is mainly governed by the solvation/desolvation effect (solvophobic interactions). These findings led us to design a rare solvent switchable system. In competition clipping experiments employing both BPY and TPY as the templates, exclusive formation of the BPY-based linear [2]rotaxane can be achieved in pure CDCl3, while in pure CD3CN, a 6.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 selectivity is achieved in favor of the TPY-based triply threaded [2]rotaxane. The detailed structural analysis of the two [2]rotaxanes as well as the solvent-dependent selectivity, may encourage more integrated approaches for the design of complex molecular architectures.
:1 selectivity is achieved in favor of the TPY-based triply threaded [2]rotaxane. The detailed structural analysis of the two [2]rotaxanes as well as the solvent-dependent selectivity, may encourage more integrated approaches for the design of complex molecular architectures.
Introduction
Mechanically interlocked molecules,1 such as [2]rotaxanes and [2]catenanes, first emerged as topologically interesting synthetic targets, and later evolved as the platform for novel molecular,2 supramolecular,3 and polymeric materials4 with unique architectures and functions, which have found many applications in nanomechanical devices,5 molecular memory,6 and reconfigurable nanovalves.7 For the synthesis of [2]rotaxanes, clipping of a macrocycle around a dumbbell-shaped template is one of the most convenient methods8 as it furnishes the synthesis with minimal steps, higher yields, and product specificity, thanks to the error-checking and self-sorting power endowed by molecular recognition, non-covalent templating and reversible dynamic covalent chemistry (DCC).9 Among several DCC reactions, imine chemistry is arguably the most versatile, and has garnered great interest in the assembly of novel structures,9e,f including molecular cages,10 Borromean rings,11 suitanes,12 catenanes,13 rotaxanes,14 and helixes.15We have been motivated by a π-guest templating protocol for the assembly of C3-symmetric macrobicycle-based interlocked nanostructures employing dynamic imine chemistry. In this regard, 2-terminal14c and 3-terminal [2]rotaxanes14g can be obtained in high yields from the reaction between simple precursors such as 1,3,5-benzenetrisaldehyde (1) and 2,2′-(ethylenedioxy)diethylamine (2) and the respective dumbbell components, such as bipyridinium (BPY) or trispyridinium (TPY) π-cationic species (Scheme 1). As shown previously by modeling14c or single crystal X-ray structures,14g the linear [2]rotaxane (LR) or the triply-threaded [2]rotaxane (TR) are stabilized by favorable aromatic–aromatic interactions between the guest and the C3-symmetric trisiminophenylene (TIP) “ceiling” and “floor”. Furthermore, the oligo(ethylene glycol) “pillars” not only provide sufficient flexibility for ideal spacing in between the two TIP units (around 7 Å apart between the floor and the ceiling), but also serve as polar binding sites to assist guest encapsulation through multiple [C–H⋯O] and [C–H⋯N] hydrogen bonding interactions. While the templating capabilities of both linear BPY and the triangular TPY guests allow us to build complex molecular architectures, more in-depth descriptions of their relative templating power are needed, which are important for answering the following questions: which clipping product is thermodynamically or kinetically favored when both BPY and TPY are subjected to the clipping reaction? And how do the weak aromatic–aromatic or [C–H⋯O] interactions respond to the surrounding solvent media? Although there are a few examples of using solvent to drive the conformational selectivity in interlocked molecules,16 examples of using solvents to affect the selectivity of products in the presence of different templates is rare.17 In this paper, we report contrasting product selectivity between the BPY and the TPY templated clipping reactions when two different solvents, namely CDCl3 and CD3CN, are used. The solvent dependence is elucidated based on 1H NMR studies and structural features are revealed by single crystal X-ray analysis of the respective linear and triply threaded interlocked molecules. We have indicated that although both clipping reactions are affected by hydrogen-bonding and aromatic–aromatic interactions in general, the nature of the aromatic–aromatic interactions is quite different. The BPY-based clipping reaction is primarily driven by electrostatic interactions between BPY and TIP surfaces, while the TPY-based reaction is mainly driven by the solvation/desolvation effect (solvophobic interactions). The different solvent responses enable us to construct an unusual solvent-driven switching system between two dynamic [2]rotaxanes in competition experiments.
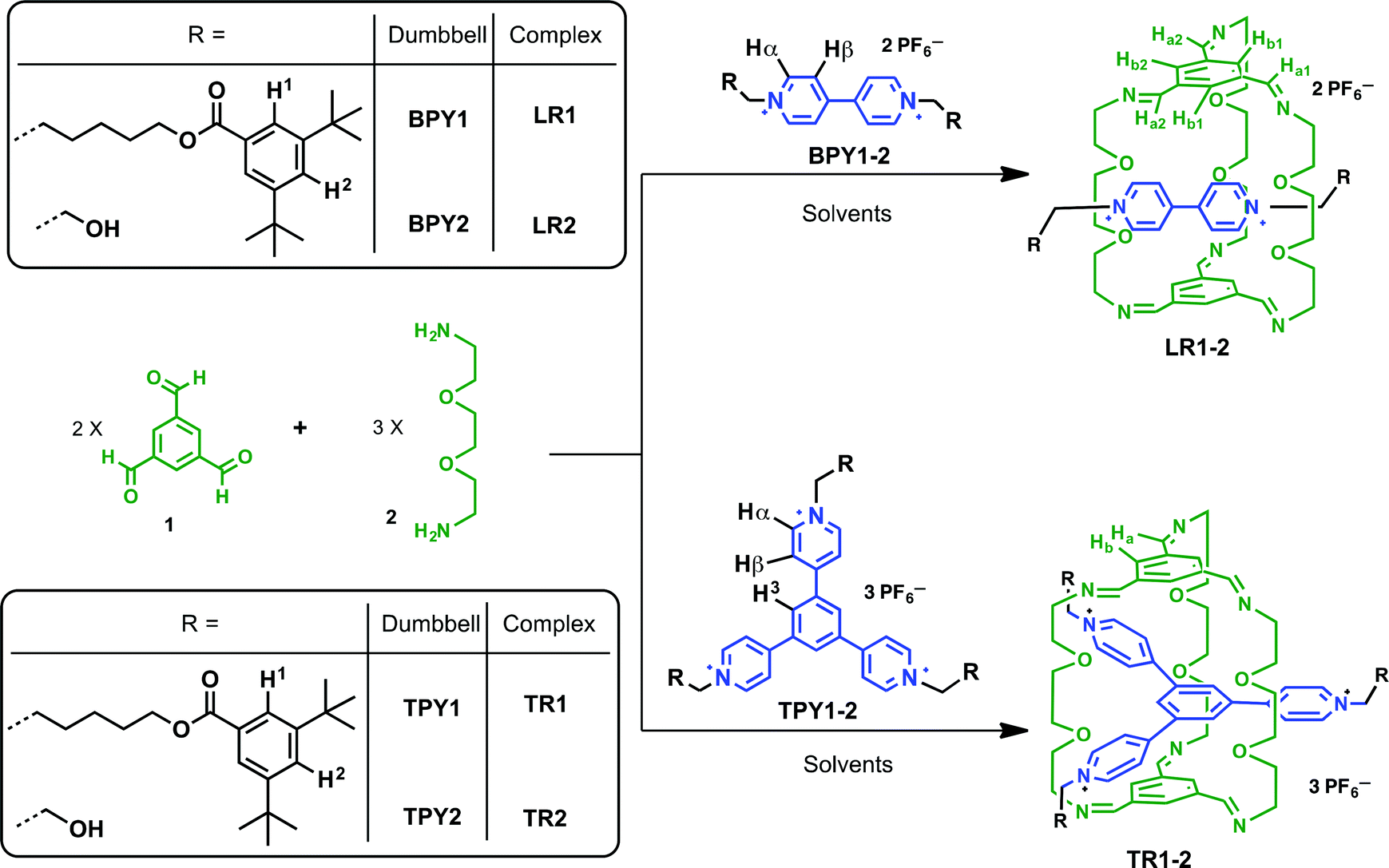 | ||
| Scheme 1 The clipping reactions of linear (LR) and triply threaded [2]rotaxanes (TR) from BPY and TPY guests. Solvents: CDCl3 and/or CD3CN. | ||
Experimental details of the clipping reactions
The clipping reactions are conducted by mixing a solution of trisaldehyde 1, diamine 2 and the corresponding BPY or TPY guest in a ratio of 2:3:1 in deuterated solvents unless noted otherwise (Scheme 1). Two different N-substituent groups are employed for both BPY and TPY compounds. The aliphatic, bulky 3,5-di-t-butylbenzoyl ester groups act as stopper units in BPY1 and TPY1 while endowing these cationic compounds with good solubility in solvents such as CD3CN and CDCl3, which is important for following the clipping reactions in single solvent systems. The short ethanol groups in BPY2 and TPY2 were introduced in order to facilitate the growth of single crystals of the respective clipping products. In the case of these ethanol derivatives, the optimized solvent systems for conducting the clipping reactions contain a mixture of CD3CN and CDCl3. It has been found that in pure CDCl3, the limited solubility of the ionic guests precludes the templating from happening, while in pure CD3CN, oligoimines formed from non-specific condensation have low solubility and quickly precipitate out of the reaction mixture after mixing, resulting in low clipping efficiency. In all cases, the clipping reactions are complete within two hours as monitored by 1H NMR spectroscopy.
When CDCl3 is used as the single solvent for BPY1 clipping reaction, LR1 is obtained as the single product, with a significant upfield shift of BPY Hα and Hβ resonances in the 1H NMR spectrum when compared with that of the free BPY1 in CDCl3 (Fig. 1a and b). In contrast to the pseudo[2]rotaxane LR2 that is in fast equilibrium with its components (Fig. S1, ESI†), the macrobicycle in LR1 is sterically hindered from slipping off the dumbbell and held in place around BPY1. Consequently, the symmetry of the macrobicyclic component is lowered so that the six imine protons and six phenylene protons become non-equivalent, each splitting into a set of two singlets in a ratio of 1:2. Increasing the amount of CD3CN to 50% and 90% while maintaining the same sample concentration dissociates LR1 by 14% and 22%, respectively, as can be seen from the appearance of free BPY1 and the macrobicyclic cage in the 1H NMR spectra (Fig. 1c and d).
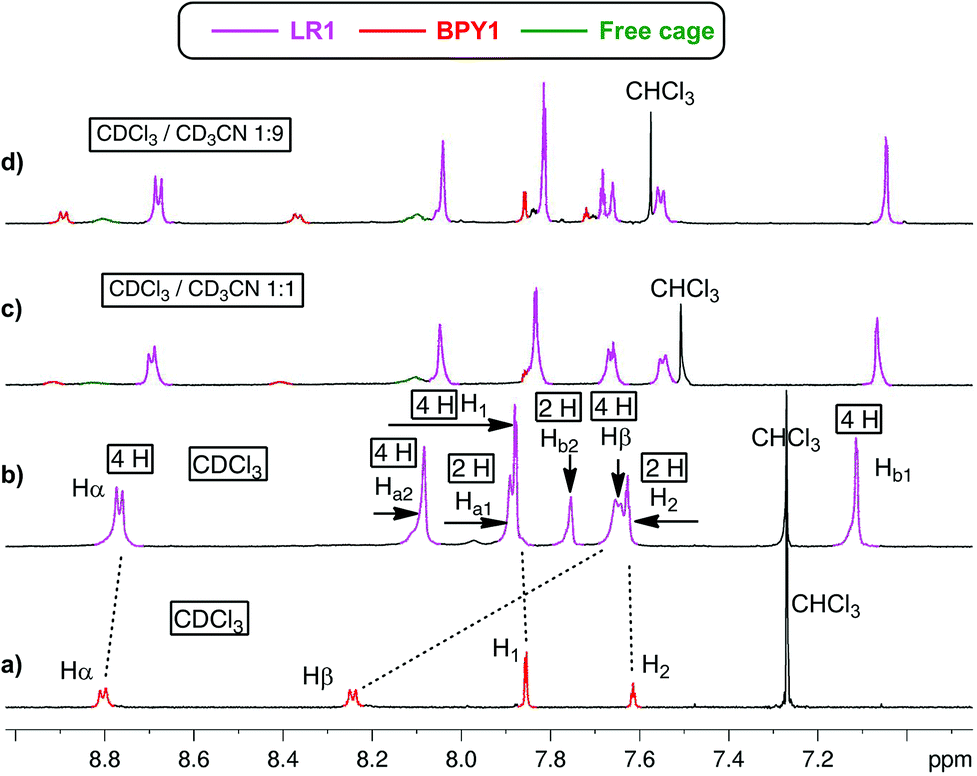 | ||
| Fig. 1 Partial 1H NMR spectra of (a) BPY1 in CDCl3, and the clipping reaction based on BPY1 in (b) CDCl3, (c) CDCl3–CD3CN (1:1, v/v), and (d) CDCl3–CD3CN (1:9, v/v). The resonances are color coded and assigned to the involved species in solution. Purple signals: LR1, red signals: BPY1, green signals: free cage. | ||
The 1H NMR spectrum of TPY1 in pure CDCl3 reveals very broad resonances of the central trispyridinium core (Fig. 2a), indicative of dynamic processes in solution. Surprisingly, when TPY1 is subjected to the clipping reaction in pure CDCl3, there is no [2]rotaxane product, and instead only TPY1 and the free macrobicyclic cage are observed (Fig. 2b). This is in sharp contrast to the same reaction that is carried out in CDCl3–CD3CN (3:5) (Fig. 2c). A new set of resonances corresponding to the formation of desired TR1 appears within 10 minutes. Significant quantities of unbound TPY1 and the free cage are also present in the solution, which, commensurate with the increasing amount of TR1, decrease as the reaction progresses and reaches equilibrium after two hours (TR1:free cage = 1:0.3). Further analysis of the 1H NMR spectrum of the interlocked species TR1 indicates that the H3 and Hβ resonances in TPY, together with the Ha and Hb resonances in TIP units, show significant upfield shifting in comparison to those of unbound TPY1 and the free cage, consistent with a mutual shielding effect between the aromatic units. In contrast, the Hα resonances of TR1 shift downfield relative to those of unbound TPY1, indicating a deshielding effect imposed by the surrounding polyimine aromatic core. Upon addition of 1.0 more equivalent of TPY1 into the reaction mixture, the equilibrium is further shifted towards nearly complete consumption of the free cage (TR1:free cage = 1:0.1, Fig. 2d). Increase of the CD3CN composition to 90% while maintaining the same TPY1 concentration further decreases the amount of free cage (TR1:free cage = 1:0.07, Fig. 2e). When this preassembled solution is evaporated and redissolved in CDCl3, the same 1H NMR spectrum as that shown in Fig. 2b is obtained, confirming the complete dissociation of TR1 in CDCl3.
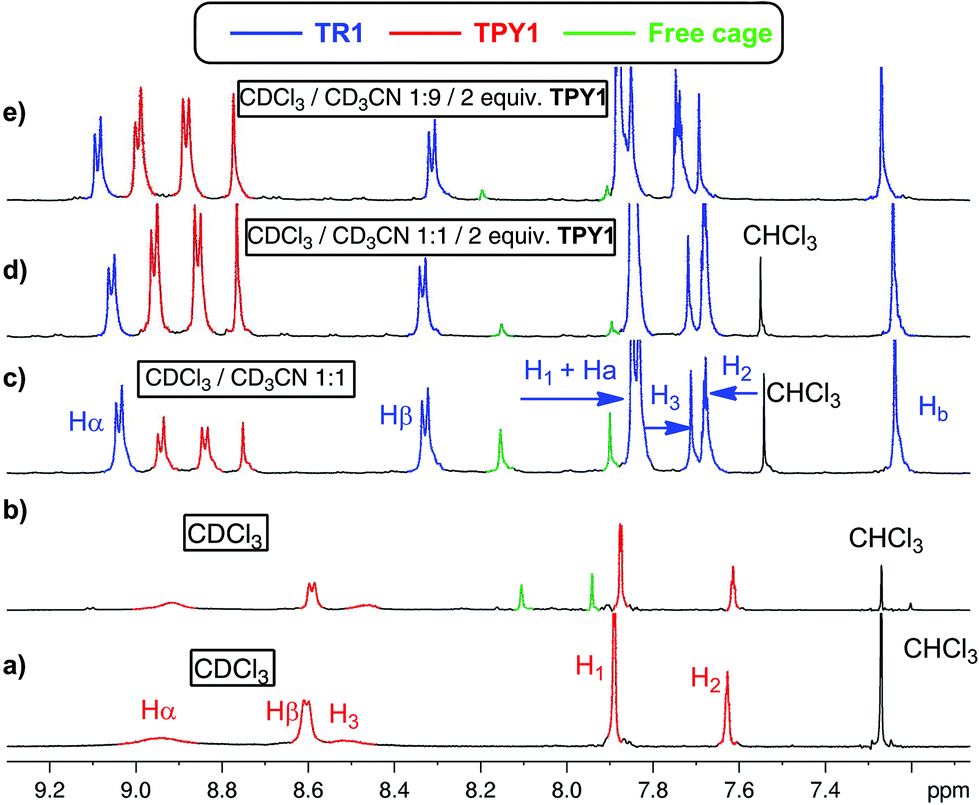 | ||
| Fig. 2 Partial 1H NMR spectra of (a) TPY1 in CDCl3, and the TPY1 based clipping reaction under different conditions. (b) 1 equiv. TPY1 clipping in CDCl3. (c) 1 equiv. TPY1 clipping in CDCl3–CD3CN (3:5, v/v). (d) 2 equiv. TPY1 clipping in CDCl3–CD3CN (3:5, v/v). (e) 2 equiv. TPY1 clipping in CDCl3–CD3CN (1:9, v/v). The resonances are color coded and assigned to the involved species in solution. Blue signals: TR1, red signals: TPY1, green signals: free cage. | ||
The contrasting solvent response prompts us to look into the detailed structural features of the respective interlocked assemblies. X-ray quality single crystals were obtained for these interlocked LR2 and TR2 using ethanol-derived BPY2 and TPY2.18 The unit cell of LR2 contains two crystallographically independent molecules, each comprising nearly parallel stacking of the BPY unit and the TIP units in the ceiling and the floor of the macrobicycle (Fig. 3). The six imine groups in the cage are coplanar with the conjugating phenylene to give two extended aromatic ring systems, while the BPY units in the two crystallographically independent molecules are twisted with dihedral angles of 7.7 and 22.6°, respectively. The distances between the centroids of TIP units to the mean plane of BPY units are all within 3.23 to 3.38 Å. The conformation is stabilized by multiple [C–H⋯O] and [C–H⋯N] hydrogen bonds between (1) two Hαs of the BPY unit and the oxygen atoms on the two nearby ethylene glycol loops on the back, (2) two Hβs of the BPY unit and two oxygen atoms on the front ethylene glycol loop, (3) methylene protons next to BPY and oxygen atoms on the nearby ethylene glycol loops, and (4) one of the hydroxyl protons and one of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N nitrogen atoms. In addition, adjacent LR2 molecules stack into extended π-stacking columns.
N nitrogen atoms. In addition, adjacent LR2 molecules stack into extended π-stacking columns.
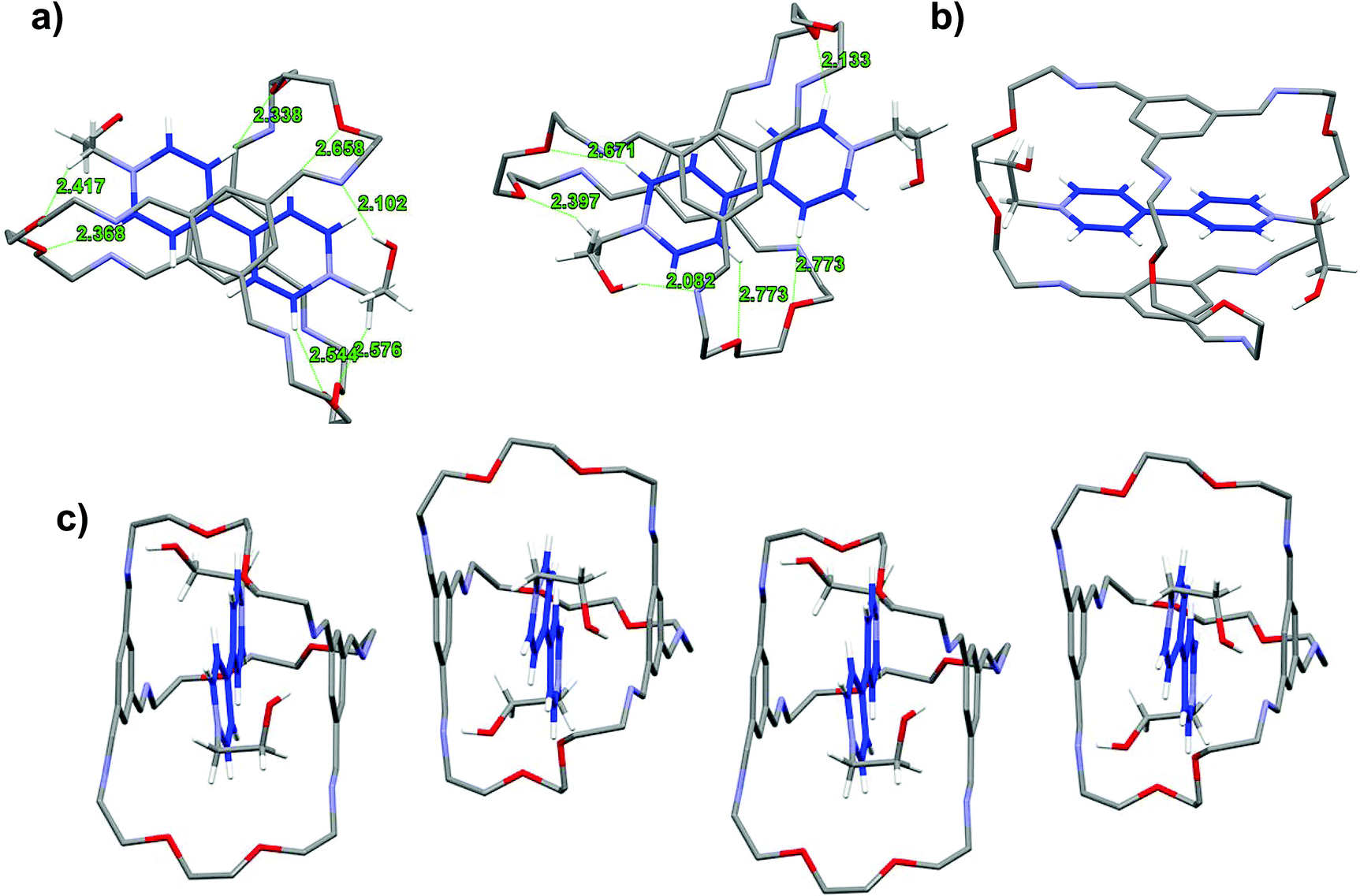 | ||
| Fig. 3 Capped stick representation of the X-ray structures of LR2. (a) Top view of X-ray structure of LR2 showing two crystallographically independent molecules in the lattice. The green dashed lines indicate [C–H⋯O] interactions. (b) Side view showing one of the two independent molecules of LR2. (c) Elongated π-stacking in the solid state. Solvent molecules, anions and hydrogen atoms on the macrobicycle are omitted for clarity. | ||
In the solid state structure of TR2 (Fig. 4),19 the TPY2 guest is sandwiched within the cavity of the macrobicyclic cage, with the pyridinium arms threading through the three oligo(ethylene glycol) orifices. The two TIP units lie nearly parallel with respect to the central phenylene ring in the TPY unit with centroid-to-plane distances of 3.50 and 3.48 Å, respectively, which are larger than those in LR2. All three pyridinium units of TPY are twisted out of the plane of its central benzene ring. The twisting satisfies the desirable geometric arrangement for multiple [C–H⋯O] interactions between the oxygen atoms of the oligo(ethylene glycol) units in the cage and four of the Hβs of the three pyridinium units in TPY. Additional [C–H⋯N] and [C–H⋯O] interactions are also observed between one of the pyridinium Hβ protons and the nitrogen atoms of a TIB unit, and between the central phenylene ring in TPY and the oxygen atoms of the oligo(ethylene glycol) units, respectively, all of which contribute collectively to the stabilization of the complex. It is worth noting that an uncomplexed TPY2 guest stacks alongside with the macrobicyclic cage to give a 1:2 host–guest complex that extends the π stacking in the solid state.
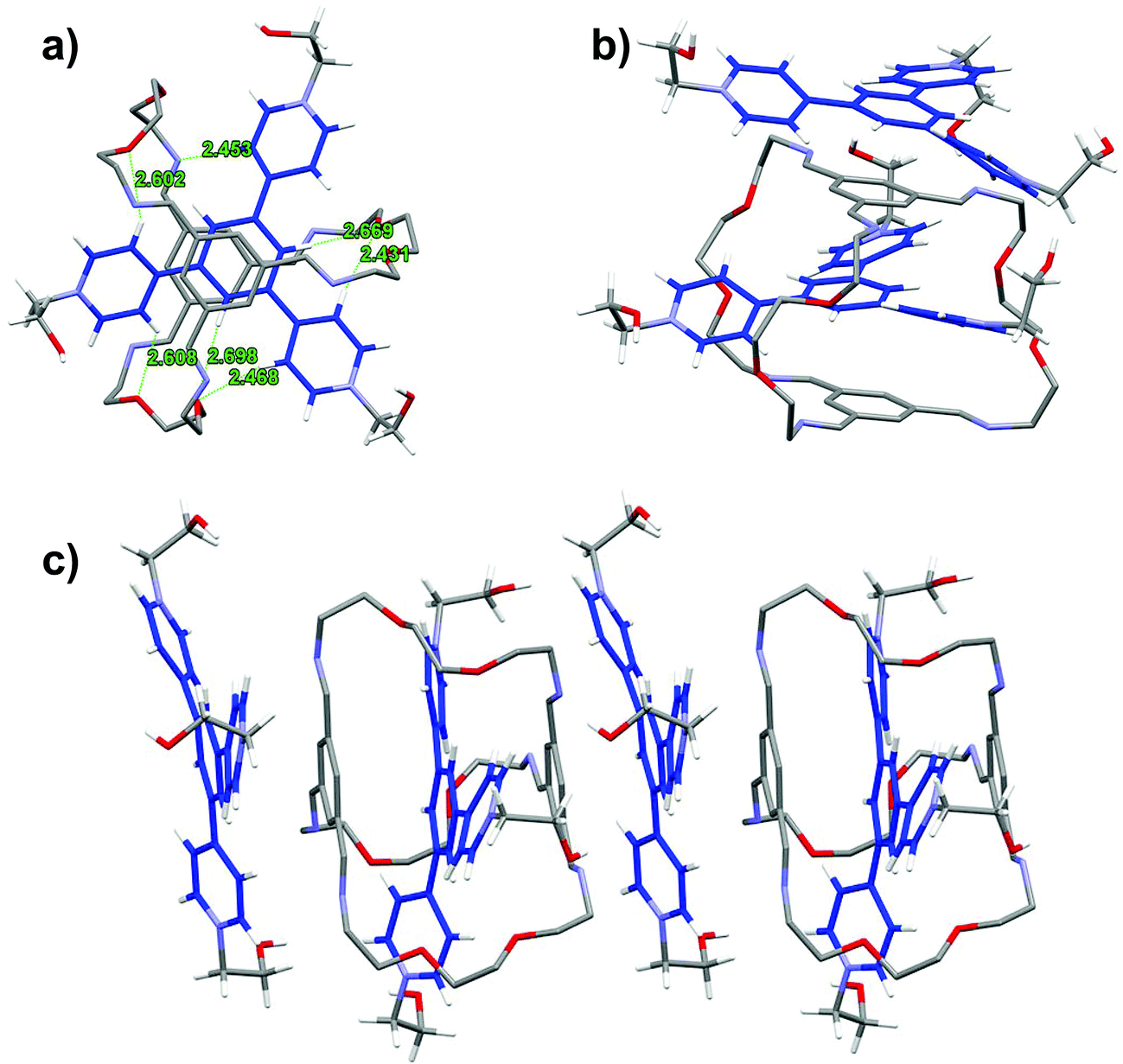 | ||
| Fig. 4 Capped stick representation of the X-ray structures of TR2. (a) Top view of the X-ray structure of TR2. The green dashed lines indicate [C–H⋯O] and [C–H⋯N] interactions. (b) Side view of the 1:2 host–guest complex structure of TR2. (c) Elongated π-stacking in the 1:2 host–guest complex in the solid state. Solvent molecules, anions and hydrogen atoms on the macrobicycle are omitted for clarity. | ||
A comparison of the structures of LR2 and TR2 indicates a similar conformation adopted by the macrobicycle; however, the relative positions of pyridinium units within the cavity are significantly different. The pyridinium units in TPY extend relatively further out of the cavity because of the central phenylene ring “spacer” and thus have no π-overlap with the TIP units on the cage, while those of the BPY have more buried π-surfaces that are overlapping with the TIP aromatic surfaces. The difference in positioning of the pyridinium units with respect to the macrobicycle also accounts for the different involvement of Hα and Hβs in hydrogen bonding interactions: both Hαs and Hβs in BPY are involved in [C–H⋯O] interactions, while in TPY, Hβs and the central phenylene protons are involved but not Hαs, the latter being distant from the hydrogen bonding acceptors on the oligo(ethylene glycol) pillars.
Competition and solvent-induced switching experiments
Encouraged by the opposite solvent responsiveness, we conducted a series of competition and solvent-driven switching experiments (Scheme 2). When equal equivalents of BPY1 and TPY1 are mixed and subjected to the clipping reaction in CDCl3, LR1 is formed exclusively, with free TPY1 in the solution (Fig. 5a). When equal volume of CD3CN is added to the mixture, TR1 started to appear and ended up with a TR1/LR1 ratio of 1.3:1 after equilibrium (Fig. 5b). The same distribution of TR1 and LR1 is observed when the clipping is conducted in a 1:1 mixture of CDCl3 and CD3CN, excluding any kinetic selection effect and confirming that a thermodynamic equilibrium is reached. Raising the composition of CD3CN to 90% and 95% increases the TR1/LR1 selectivity to 5.1:1 and 6.7:1, respectively (Fig. 5c and d). The solvent-dependent selectivity is summarized in Scheme 2.
 | ||
| Scheme 2 Scheme of the competition experiment and the corresponding solvent-dependent selectivity. | ||
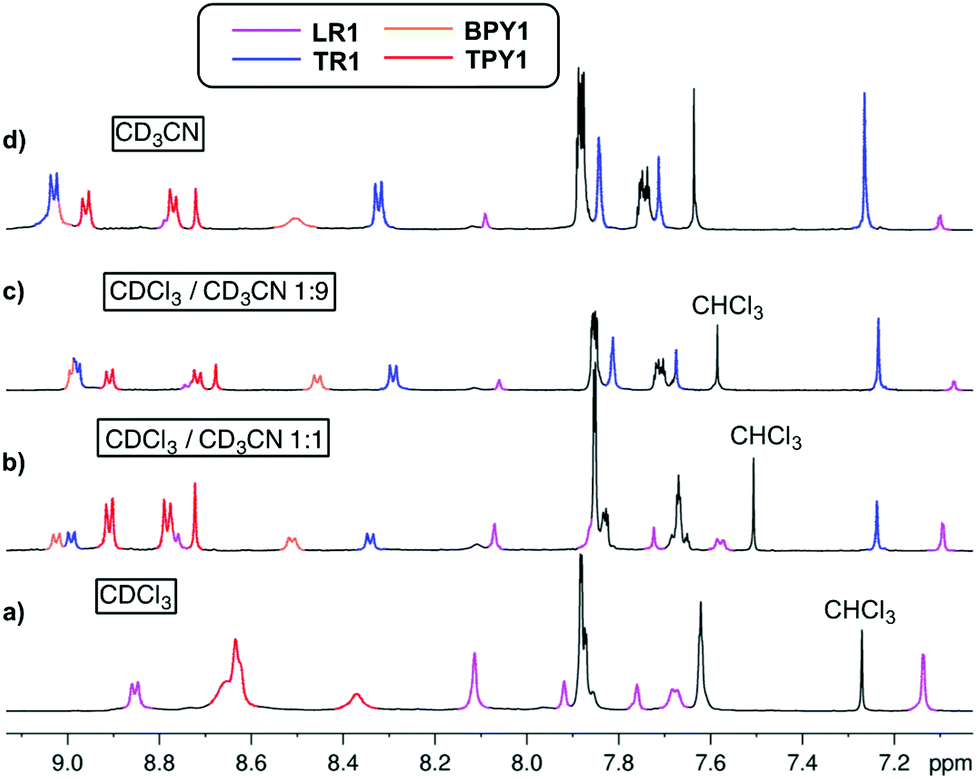 | ||
| Fig. 5 Competition clipping experiment conducted in (a) CDCl3, (b) CDCl3–CD3CN (1:1), (c) CDCl3–CD3CN (1:9) and (d) CDCl3–CD3CN (1:19). Non-overlapping resonances are color coded and assigned to either the templates or the [2]rotaxanes. Purple signals: LR1, blue signals: TR1, orange signals: BPY1, red signals: TPY1. | ||
Discussion of the solvent effect
The main driving forces involved in these [2]rotaxanes are aromatic–aromatic and hydrogen bonding interactions. Polar solvents weaken the hydrogen bonding interactions. For the impact of solvent polarity on aromatic–aromatic interactions, since the nature of such interactions is complementary electrostatic interactions and/or solvation/desolvation effects (i.e. solvophobic interactions),20 the impact is ambipolar: higher polarity weakens electrostatic interactions, and promotes solvophobic interactions in between π surfaces.For LR1, the addition of more polar CD3CN into CDCl3 decomplexes the interlocked structure, suggesting that the energy gain from aromatic–aromatic interactions, if any, is inadequate to compensate for the destabilized hydrogen bonding interactions. In the case of TR1, the solvent response is the opposite. Increasing the composition of CD3CN in the solvent system favors the formation of TR1, while CDCl3 decomplexes the interlocked [2]rotaxane, and in 100% CDCl3 there is no TR1 despite the fact that it is a more benign solvent for hydrogen bonding. While this suggests that aromatic–aromatic interactions are more dominant than [C–H⋯O] hydrogen bonding interactions in stabilizing the interlocked structure, there might exist other competing processes that affect the equilibrium, as indicated by the broad 1H NMR resonances of the aromatic core of TPY1 in CDCl3. Variable temperature experiments are conducted to reveal the temperature dependence of the peak broadening. As shown in Fig. S2 in ESI,† the Hα and H3 resonances of the TPY1 core are shifted and become broader as the temperature is lowered, and are almost concealed in the baseline as the temperature approaches the melting point of CDCl3. The line broadening implies that the resonances are close to coalescence between equilibrating species, which is presumably a result of self-aggregation of TPY1 through dimerization/oligomerization. It is postulated that while the lipophilic end groups of TPY1 ensure good solubility in less polar solvents like CDCl3, the tricationic core of TPY1 is poorly solvated. Consequently, TPY1 molecules tend to aggregate with the trispyridinium units congested together to form an inner core with a surrounding outer shell of the alkyl ester end groups. In less polar CDCl3, this aggregation gives the least exposed polar surface area, and is more favored over the formation of interlocked species. In contrast, well-resolved resonances are observed for TPY1 in the presence of CD3CN, (Fig. S8 in ESI†) indicating better solvation of the trispyridinium core and insignificant aggregation. In addition, the better yield of TR1 at higher composition of CD3CN suggests that while aggregation and [C–H⋯O] hydrogen bonding interactions become insignificant, aromatic–aromatic interactions are reinforced to compensate for the energy penalty. The different solvent responses can be further discussed on account of the following aspects:
(1) The [C–H⋯O] hydrogen bonding strength is different. In BPY1, both Hα and Hβ-protons are involved in [C–H⋯O] interactions, while in TPY1, only Hβ-protons and the phenylene protons are involved in [C–H⋯O] interactions, which are much less acidic than Hα and weaker H-bonding donors.
(2) The electron densities of the guest π-surfaces are different. Electrostatic surface potential (ESP) plots indicate (Fig. S3 in ESI†) that both BPY and TPY aromatic cores are electron deficient while the TIP units of the macrobicycle are relatively electron rich. Cyclic voltammetric studies performed in MeCN (Fig. S4 in ESI†) indicate that BPY1 has a much less negative half-wave reductive potential than TPY1 (E1/2: −0.81 V vs. −1.38 V, with reference to Fc/Fc+), confirming that BPY1 is a stronger electron acceptor than TPY1. Despite TPY1's weaker electron accepting ability, TR1 is selectively formed in CD3CN over LR1 (6.7:1), suggesting that there are other driving forces than electrostatic interaction that account for TPY's better templating efficiency over BPY in a polar solvent. The weaker electrostatic interaction in TR1 is also supported by its solid-state structure, in which the most electron deficient parts of TPY, i.e. pyridinium units, have barely any π-overlap with the TIP units.
(3) The size and charge of π-surfaces are different. The cationic pyridinium unit is better solvated in more polar solvent such as MeCN. BPY1 contains two pyridinium units while TPY1 has a larger π-surface with three pyridinium units and one neutral central phenylene ring. In less polar CDCl3, TPY1 experiences significant aggregation due to poor solvation of the charged π-surface. In more polar CD3CN, the interlocked structure is favored by situating the TPY core inside the cavity of the macrobicyclic cage with three pyridinium units sticking out, and the central phenylene ring overlapping with the two TIP π-surfaces. This geometry ensures both sufficient solvation of the cationic pyridinium units and effective shielding of the central phenylene unit to preserve the buried hydrophobic surface area from unfavorable interactions with polar solvent. The formation of a 1:2 complex in the solid-state structure of TR2 is probably also driven by such a solvation effect.
Overall, the BPY templated assembly of linear [2]rotaxanes is driven by a combination of hydrogen bonding interactions and complementary electrostatics but not so much by solvophobic interactions. In the cases of TPY templated assembly of triply threaded [2]rotaxanes, the complexation is driven by a combination of hydrogen bonding and solvophobic interactions, but not much by complementary electrostatics. It is the difference in collective non-covalent interactions that accounts for the opposite selectivity in different solvents.
Conclusions
In summary, we have demonstrated the solvent switchable formation of two dynamic [2]rotaxanes when two π cationic guests are employed. The solvent-dependent selectivity relies on the subtle differences of the aromatic–aromatic interactions that govern the templated formation of two [2]rotaxanes. The BPY-based clipping reaction is driven by electrostatic interactions between aromatic surfaces, while the TPY-based reaction is driven by solvophobic interactions. In competition clipping experiments employing both BPY and TPY as the templates, exclusive formation of the BPY-based linear [2]rotaxane can be achieved in pure CDCl3, while in pure CD3CN, a 8:1 selectivity is achieved with the TPY-based triply threaded [2]rotaxane as the major product. Combining the structural features of the two [2]rotaxanes and the high solvent-dependent selectivity provides a unique test bed for probing the nature of aromatic–aromatic interactions, which can be essential for the design of complex molecular architectures that greatly rely on weak but cooperative non-covalent interactions.
Acknowledgements
This study was performed at the Molecular Foundry and Advanced Light Source, Lawrence Berkeley National Laboratory, supported by the Office of Science, Office of Basic Energy Sciences, Scientific User Facilities Division, of the U.S. Department of Energy under contract no. DE-AC02-05CH11231.Notes and references
- (a) G. Barin, A. Coskun, M. M. G. Fouda and J. F. Stoddart, ChemPlusChem, 2012, 77, 159–185 CrossRef CAS; (b) C. J. Bruns and J. F. Stoddart, Top. Curr. Chem., 2012, 323, 19–72 CrossRef CAS.
- (a) Y. Luo, C. P. Collier, J. O. Jeppesen, K. A. Nielsen, E. DeIonno, G. Ho, J. Perkins, H. R. Tseng, T. Yamamoto, J. F. Stoddart and J. R. Heath, ChemPhysChem, 2002, 3, 519–525 CrossRef CAS; (b) S. A. Vignon, T. Jarrosson, T. Iijima, H. R. Tseng, J. K. M. Sanders and J. F. Stoddart, J. Am. Chem. Soc., 2004, 126, 9884–9885 CrossRef CAS PubMed; (c) J. Berna, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Perez, P. Rudolf, G. Teobaldi and F. Zerbetto, Nat. Mater., 2005, 4, 704–710 CrossRef CAS PubMed; (d) D. A. Leigh, M. A. F. Morales, E. M. Perez, J. K. Y. Wong, C. G. Saiz, A. M. Z. Slawin, A. J. Carmichael, D. M. Haddleton, A. M. Brouwer, W. J. Buma, G. W. H. Wurpel, S. Leon and F. Zerbetto, Angew. Chem., Int. Ed., 2005, 44, 3062–3067 CrossRef CAS PubMed; (e) T. D. Nguyen, Y. Liu, S. Saha, K. C. F. Leung, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2007, 129, 626–634 CrossRef CAS PubMed; (f) V. Serreli, C.-F. Lee, E. R. Kay and D. A. Leigh, Nature, 2007, 445, 523–527 CrossRef CAS PubMed; (g) E. Coronado, P. Gavina and S. Tatay, Chem. Soc. Rev., 2009, 38, 1674–1689 RSC; (h) F. Durola, J. Lux and J.-P. Sauvage, Chem.–Eur. J., 2009, 15, 4124–4134 CrossRef CAS PubMed; (i) S. Silvi, M. Venturi and A. Credi, J. Mater. Chem., 2009, 19, 2279–2294 RSC; (j) Y. Jiang, J.-B. Guo and C.-F. Chen, Org. Lett., 2010, 12, 4248–4251 CrossRef CAS PubMed; (k) C.-F. Chen, Chem. Commun., 2011, 47, 1674–1688 RSC; (l) S.-Z. Hu and C.-F. Chen, Chem.–Eur. J., 2011, 17, 5424–5431 CrossRef CAS PubMed.
- (a) K. Kim, Chem. Soc. Rev., 2002, 31, 96–107 RSC; (b) V. Sindelar, S. Silvi and A. E. Kaifer, Chem. Commun., 2006, 2185–2187 RSC; (c) F. Huang, D. S. Nagvekar, X. Zhou and H. W. Gibson, Macromolecules, 2007, 40, 3561–3567 CrossRef CAS; (d) F. Wang, C. Han, C. He, Q. Zhou, J. Zhang, C. Wang, N. Li and F. Huang, J. Am. Chem. Soc., 2008, 130, 11254–11255 CrossRef CAS PubMed; (e) H. Zhang, Q. Wang, M. Liu, X. Ma and H. Tian, Org. Lett., 2009, 11, 3234–3237 CrossRef CAS PubMed; (f) F. Wang, J. Zhang, X. Ding, S. Dong, M. Liu, B. Zheng, S. Li, L. Wu, Y. Yu, H. W. Gibson and F. Huang, Angew. Chem., Int. Ed., 2010, 49, 1090–1094 CrossRef CAS PubMed; (g) S. Dong, Y. Luo, X. Yan, B. Zheng, X. Ding, Y. Yu, Z. Ma, Q. Zhao and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1905–1909 CrossRef CAS PubMed; (h) Z. Niu, F. Huang and H. W. Gibson, J. Am. Chem. Soc., 2011, 133, 2836–2839 CrossRef CAS PubMed; (i) Z. Zhang, Y. Luo, J. Chen, S. Dong, Y. Yu, Z. Ma and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1397–1401 CrossRef CAS PubMed; (j) S. Dong, X. Yan, B. Zheng, J. Chen, X. Ding, Y. Yu, D. Xu, M. Zhang and F. Huang, Chem.–Eur. J., 2012, 18, 4195–4199 CrossRef CAS PubMed; (k) X. Yan, D. Xu, X. Chi, J. Chen, S. Dong, X. Ding, Y. Yu and F. Huang, Adv. Mater., 2012, 24, 362–369 CrossRef CAS PubMed; (l) B. Zheng, F. Wang, S. Dong and F. Huang, Chem. Soc. Rev., 2012, 41, 1621–1636 RSC.
- (a) P. N. Taylor, M. J. O'Connell, L. A. McNeill, M. J. Hall, R. T. Aplin and H. L. Anderson, Angew. Chem., Int. Ed., 2000, 39, 3456–3460 CrossRef CAS; (b) H. W. Gibson, Z. Ge, F. Huang, J. W. Jones, H. Lefebvre, M. J. Vergne and D. M. Hercules, Macromolecules, 2005, 38, 2626–2637 CrossRef CAS; (c) Y. Jiang, J. Wu, L. He, C. Tu, X. Zhu, Q. Chen, Y. Yao and D. Yan, Chem. Commun., 2008, 6351–6353 RSC; (d) Q. Li, W. Zhang, O. S. Miljanic, C.-H. Sue, Y.-L. Zhao, L. Liu, C. B. Knobler, J. F. Stoddart and O. M. Yaghi, Science, 2009, 325, 855–859 CrossRef CAS PubMed; (e) C. Travelet, P. Hebraud, C. Perry, C. Brochon, G. Hadziioannou, A. Lapp and G. Schlatter, Macromolecules, 2010, 43, 1915–1921 CrossRef CAS; (f) N. Momcilovic, P. G. Clark, A. J. Boydston and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 19087–19089 CrossRef CAS PubMed; (g) A. Coskun, M. Hmadeh, G. Barin, F. Gandara, Q. Li, E. Choi, N. L. Strutt, D. B. Cordes, A. M. Z. Slawin, J. F. Stoddart, J.-P. Sauvage and O. M. Yaghi, Angew. Chem., Int. Ed., 2012, 51, 2160–2163 CrossRef CAS PubMed; (h) M. Zhang, D. Xu, X. Yan, J. Chen, S. Dong, B. Zheng and F. Huang, Angew. Chem., Int. Ed., 2012, 51, 7011–7015 CrossRef CAS PubMed.
- (a) T. J. Huang, B. Brough, C. M. Ho, Y. Liu, A. H. Flood, P. A. Bonvallet, H. R. Tseng, J. F. Stoddart, M. Baller and S. Magonov, Appl. Phys. Lett., 2004, 85, 5391–5393 CrossRef CAS PubMed; (b) Y. Liu, A. Flood, R. Moskowitz and J. Stoddart, Chem.–Eur. J., 2005, 11, 369–385 CrossRef PubMed; (c) B. K. Juluri, A. S. Kumar, Y. Liu, T. Ye, Y.-W. Yang, A. H. Flood, L. Fang, J. F. Stoddart, P. S. Weiss and T. J. Huang, ACS Nano, 2009, 3, 291–300 CrossRef CAS PubMed.
- (a) E. W. Wong, C. P. Collier, M. Behloradsky, F. M. Raymo, J. F. Stoddart and J. R. Heath, J. Am. Chem. Soc., 2000, 122, 5831–5840 CrossRef CAS; (b) C. P. Collier, J. O. Jeppesen, Y. Luo, J. Perkins, E. W. Wong, J. R. Heath and J. F. Stoddart, J. Am. Chem. Soc., 2001, 123, 12632–12641 CrossRef CAS PubMed; (c) A. R. Pease, J. O. Jeppesen, J. F. Stoddart, Y. Luo, C. P. Collier and J. R. Heath, Acc. Chem. Res., 2001, 34, 433–444 CrossRef CAS PubMed; (d) A. H. Flood, J. F. Stoddart, D. W. Steuerman and J. R. Heath, Science, 2004, 306, 2055–2056 CrossRef CAS PubMed; (e) J. W. Choi, A. H. Flood, D. W. Steuerman, S. Nygaard, A. B. Braunschweig, N. N. P. Moonen, B. W. Laursen, Y. Luo, E. DeIonno, A. J. Peters, J. O. Jeppesen, K. Xu, J. F. Stoddart and J. R. Heath, Chem.–Eur. J., 2005, 12, 261–279 CrossRef PubMed; (f) J. E. Green, J. W. Choi, A. Boukai, Y. Bunimovich, E. Johnston-Halperin, E. DeIonno, Y. Luo, B. A. Sheriff, K. Xu, Y. S. Shin, H.-R. Tseng, J. F. Stoddart and J. R. Heath, Nature, 2007, 445, 414–417 CrossRef CAS PubMed; (g) W. Zhang, E. DeIonno, W. R. Dichtel, L. Fang, A. Trabolsi, J.-C. Olsen, D. Benitez, J. R. Heath and J. F. Stoddart, J. Mater. Chem., 2011, 21, 1487–1495 RSC.
- (a) R. Hernandez, H.-R. Tseng, J. W. Wong, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2004, 126, 3370–3371 CrossRef CAS PubMed; (b) T. D. Nguyen, H.-R. Tseng, P. C. Celestre, A. H. Flood, Y. Liu, J. F. Stoddart and J. I. Zink, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10029–10034 CrossRef CAS PubMed; (c) S. Angelos, E. Johansson, J. F. Stoddart and J. I. Zink, Adv. Funct. Mater., 2007, 17, 2261–2271 CrossRef CAS; (d) T. D. Nguyen, Y. Liu, S. Saha, K. C. F. Leung, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2007, 129, 626–634 CrossRef CAS PubMed; (e) S. Angelos, Y.-W. Yang, K. Patel, J. F. Stoddart and J. I. Zink, Angew. Chem., Int. Ed., 2008, 47, 2222–2226 CrossRef CAS PubMed; (f) H. Meng, M. Xue, T. Xia, Y.-L. Zhao, F. Tamanoi, J. F. Stoddart, J. I. Zink and A. E. Nel, J. Am. Chem. Soc., 2010, 132, 12690–12697 CrossRef CAS PubMed.
- (a) P. L. Anelli, P. R. Ashton, R. Ballardini, V. Balzani, M. Delgado, M. T. Gandolfi, T. T. Goodnow, A. E. Kaifer, D. Philp, M. Pietraszkiewicz, L. Prodi, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, J. Am. Chem. Soc., 1992, 114, 193–218 CrossRef CAS; (b) J. Berna, G. Bottari, D. A. Leigh and E. M. Perez, Pure Appl. Chem., 2007, 79, 39–54 CrossRef CAS.
- (a) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef; (b) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed; (c) B. H. Northrop, F. Arico, N. Tangchiavang, J. D. Badjic and J. F. Stoddart, Org. Lett., 2006, 8, 3899–3902 CrossRef CAS PubMed; (d) C. D. Meyer, C. S. Joiner and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 1705–1723 RSC; (e) M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC; (f) Y. Liu and Z.-T. Li, Aust. J. Chem., 2013, 66, 9–22 CrossRef CAS.
- (a) O. Francesconi, A. Ienco, G. Moneti, C. Nativi and S. Roelens, Angew. Chem., Int. Ed., 2006, 45, 6693–6696 CrossRef CAS PubMed; (b) X. Liu, Y. Liu, G. Li and R. Warmuth, Angew. Chem., Int. Ed., 2006, 45, 901–904 CrossRef CAS PubMed; (c) X. Liu and R. Warmuth, J. Am. Chem. Soc., 2006, 128, 14120–14127 CrossRef CAS PubMed; (d) J. Luo, T. Lei, X. Xu, F.-M. Li, Y. Ma, K. Wu and J. Pei, Chem.–Eur. J., 2008, 14, 3860–3865 CrossRef CAS PubMed; (e) D. Xu and R. Warmuth, J. Am. Chem. Soc., 2008, 130, 7520–7521 CrossRef CAS PubMed; (f) T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed; (g) X.-N. Xu, L. Wang and Z.-T. Li, Chem. Commun., 2009, 6634–6636 RSC; (h) X.-N. Xu, L. Wang, G.-T. Wang, J.-B. Lin, G.-Y. Li, X.-K. Jiang and Z.-T. Li, Chem.–Eur. J., 2009, 15, 5763–5774 CrossRef CAS PubMed; (i) R. A. Bilbeisi, J. K. Clegg, N. Elgrishi, X. d. Hatten, M. Devillard, B. Breiner, P. Mal and J. R. Nitschke, J. Am. Chem. Soc., 2011, 134, 5110–5119 CrossRef PubMed; (j) A. Granzhan, C. M. Schouwey, T. Riis-Johannessen, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2011, 133, 7106–7115 CrossRef CAS PubMed; (k) Y. Jin, B. A. Voss, A. Jin, H. Long, R. D. Noble and W. Zhang, J. Am. Chem. Soc., 2011, 133, 6650–6658 CrossRef CAS PubMed; (l) J. Sun, J. L. Bennett, T. J. Emge and R. Warmuth, J. Am. Chem. Soc., 2011, 133, 3268–3271 CrossRef CAS PubMed; (m) P. P. Neelakandan, A. Jimenez and J. R. Nitschke, Chem. Sci., 2014 10.1039/C3SC53172D.
- (a) K. S. Chichak, S. J. Cantrill, A. R. Pease, S. H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS PubMed; (b) C. D. Meyer, R. S. Forgan, K. S. Chichak, A. J. Peters, N. Tangchaivang, G. W. V. Cave, S. I. Khan, S. J. Cantrill and J. F. Stoddart, Chem.–Eur. J., 2010, 16, 12570–12581 CrossRef CAS PubMed.
- A. R. Williams, B. H. Northrop, T. Chang, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 2006, 45, 6665–6669 CrossRef CAS PubMed.
- (a) T. Hasell, X. F. Wu, J. T. A. Jones, J. Bacsa, A. Steiner, T. Mitra, A. Trewin, D. J. Adams and A. I. Cooper, Nat. Chem., 2010, 2, 750–755 CrossRef CAS PubMed; (b) G. Koshkakaryan, D. Cao, L. M. Klivansky, S. J. Teat, J. L. Tran and Y. Liu, Org. Lett., 2010, 12, 1528–1531 CrossRef CAS PubMed; (c) S. P. Black, A. R. Stefankiewicz, M. M. J. Smulders, D. Sattler, C. A. Schalley, J. R. Nitschke and J. K. M. Sanders, Angew. Chem., Int. Ed., 2013, 52, 5749–5752 CrossRef CAS PubMed; (d) S.-T. Tung, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Angew. Chem., Int. Ed., 2013, 52, 13269–13272 CrossRef CAS PubMed.
- (a) P. T. Glink, A. I. Oliva, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 2001, 40, 1870–1875 CrossRef CAS; (b) J. Wu, K. C.-F. Leung and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17266–17271 CrossRef CAS PubMed; (c) L. M. Klivansky, G. Koshkakaryan, D. Cao and Y. Liu, Angew. Chem., Int. Ed., 2009, 48, 4185–4189 CrossRef CAS PubMed; (d) J. Yin, S. Dasgupta and J. Wu, Org. Lett., 2010, 12, 1712–1715 CrossRef CAS PubMed; (e) S. Dasgupta and J. Wu, Org. Biomol. Chem., 2011, 9, 3504–3515 RSC; (f) M. E. Belowich, C. Valente, R. A. Smaldone, D. C. Friedman, J. Thiel, L. Cronin and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 5243–5261 CrossRef CAS PubMed; (g) A. Pun, D. A. Hanifi, G. Kiel, E. O'Brien and Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 13119–13122 CrossRef CAS PubMed; (h) J. Li, P. Wei, X. Wu, M. Xue, X. Yan and Q. Zhou, RSC Adv., 2013, 3, 21289–21293 RSC.
- A. Sørensen, A. M. Castilla, T. K. Ronson, M. Pittelkow and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 11273–11277 CrossRef PubMed.
- (a) S. J. Loeb, J. Tiburcio and S. J. Vella, Chem. Commun., 2006, 1598–1600 RSC; (b) N. D. Suhan, L. Allen, M. T. Gharib, E. Viljoen, S. J. Vella and S. J. Loeb, Chem. Commun., 2011, 47, 5991–5993 RSC; (c) C. Wang, D. Cao, A. C. Fahrenbach, L. Fang, M. A. Olson, D. C. Friedman, S. Basu, S. K. Dey, Y. Y. Botros and J. F. Stoddart, J. Phys. Org. Chem., 2012, 25, 544–552 CrossRef CAS; (d) M. R. Panman, B. H. Bakker, D. den Uyl, E. R. Kay, D. A. Leigh, W. J. Buma, A. M. Brouwer, J. A. J. Geenevasen and S. Woutersen, Nat. Chem., 2013, 5, 929–934 CrossRef CAS PubMed.
- S. Zarra, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 4837–4840 CrossRef CAS PubMed.
- Single crystals are grown by diffusing diethyl ether into the solution of LR2 (in CD3CN–MeOD) and TR2 (in CD3CN–CDCl3).
- The same structure has been reported previously. The data reported here are acquired on a new set of crystals and refined separately.
- (a) C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS; (b) L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed; (c) C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 977101–977102. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3qo00074e |
| This journal is © the Partner Organisations 2014 |