Facile multi-decagram synthesis of methyl but-2-ynoate†
Benjamin
Darses‡
,
Iacovos N.
Michaelides
and
Darren J.
Dixon
*
Department of Chemistry, Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: Darren.Dixon@chem.ox.ac.uk
First published on 27th January 2014
Abstract
A high yielding and operationally simple protocol affords multi-decagram quantities of the synthetically useful methyl but-2-ynoate from commercially available starting materials and reagents.
Methyl but-2-ynoate (3) is the simplest internal alkyl alkynoate making it a molecule commonly used in numerous methodologies in order to demonstrate the scope of new reactions.1 This compound has also been used in a number of natural product total syntheses2 as well as in medicinal chemistry projects.3 In our recent efforts toward the synthesis of calyciphylline A-type Daphniphyllum alkaloids,4 multi-gram quantities of this specific methyl alkynoate were required. Although available in small quantities from various suppliers, the high price necessitated its straightforward synthesis from commercially available and inexpensive starting materials and reagents.
One of the current procedures to synthesize 3 involves a four-step protocol5 starting from the reaction of methyl bromoacetate and triphenylphosphine to give phosphane 4 after a deprotonation (Scheme 2). This is then acetylated and subsequently subjected to a technically challenging vacuum pyrolysis at 180 °C to promote an internal Wittig reaction giving 3 in a 60% overall yield. In another method, pyrazole 5 is oxidized to 3 either by using 2 equivalents of hypervalent iodine6 or 2 equivalents of the highly toxic thallium(III) nitrate7 giving 3 in 60% and 78% yield respectively (toxic Pb(OAc)4 can also be used as an alternative for this oxidation however the product is only afforded in 35% yield).8 A simpler method involves the esterification of the relatively costly 2-butynoic acid (6) in methanol giving the desired molecule in 4 days with a 67% yield after an aqueous work up.9 Finally a two-step protocol starting with the bromination of propyne gas (7)10 followed by a palladium catalyzed carbonylation of the bromopropyne11 product using CO gas (1 atm) in methanol affords the product in an overall 61% yield over two steps.12 Iodopropyne can also be carbonylated using PdI2, giving a higher 80% yield for the second step; however, iodopropyne is expensive and only available from a limited number of suppliers.
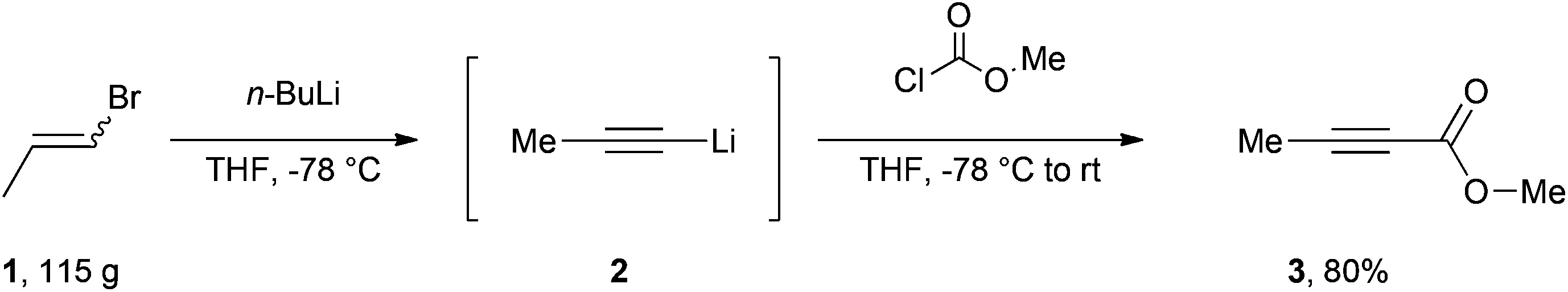 | ||
| Scheme 1 Facile synthesis of methyl but-2-ynoate. | ||
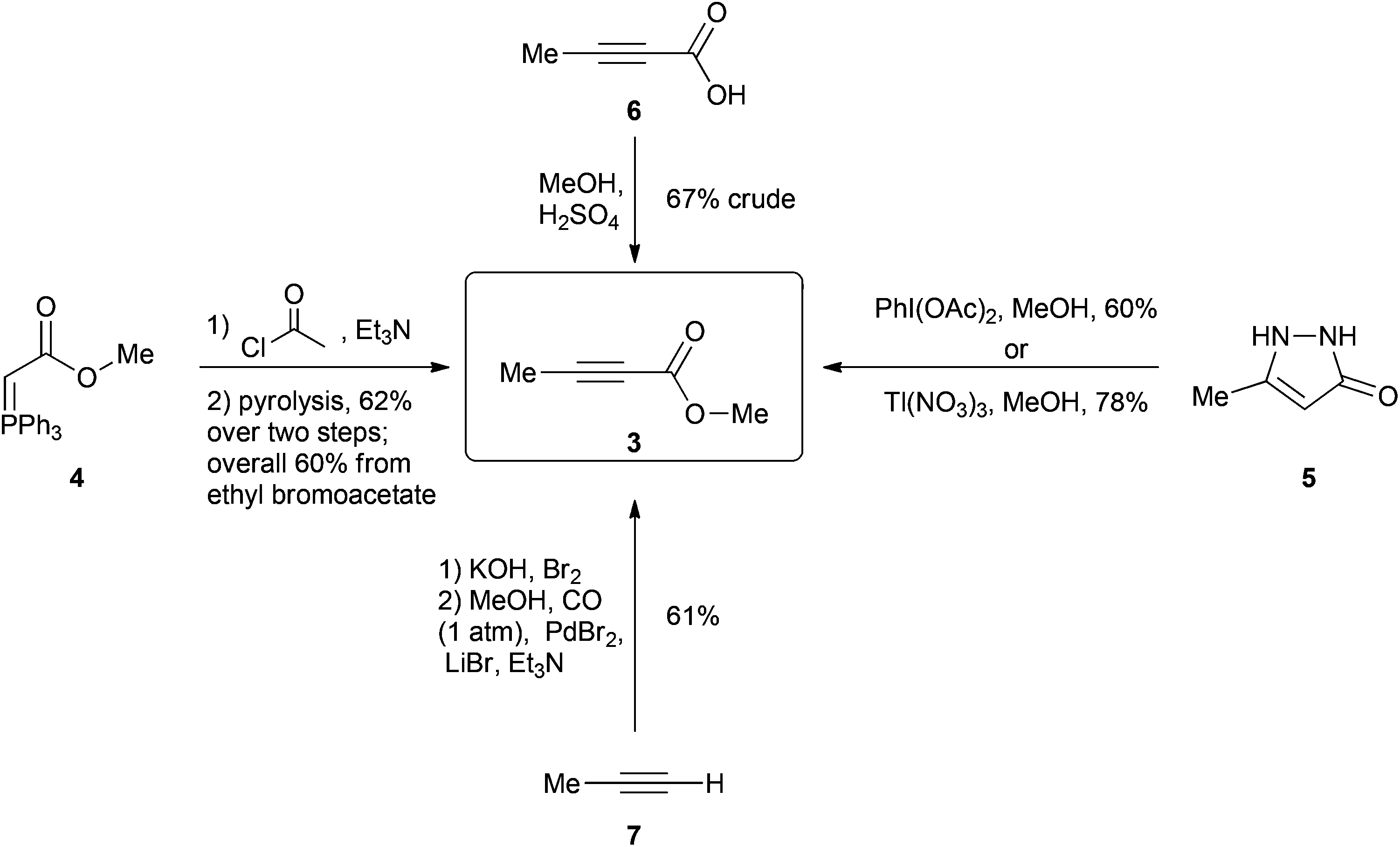 | ||
| Scheme 2 Current methods for the synthesis of 3. | ||
All the aforementioned procedures present certain drawbacks which would make the synthesis of this simple alkynoate challenging on a multi-gram scale. Our aim was to gain rapid access to large quantities of 3 which if possible, avoided the use of propyne and CO gas, long reaction periods, toxic reagents and by-products as well as the use of expensive starting materials. Furthermore, it was desirable to have a high yielding, operationally simple and reproducible experimental procedure. We therefore believed that it would be useful to devise a straightforward method to prepare large quantities of 3 efficiently, at a low-cost and with an improved yield.
Our efficient protocol (Scheme 1) is based on a method previously reported for the preparation of propargylic secondary alcohols and ketones.13 In our synthesis we make use of the cheap and commercially available mixture of Z and E isomers of 1-bromo-1-propene (1) by treating it with n-BuLi at −78 °C to generate the propynyllithium anion (2) in situ. This nucleophile is then reacted with methyl chloroformate to afford the desired molecule in 80% yield. The procedure is more atom-economic when compared to most of the existing methodologies and furthermore gives rise to LiCl and LiBr as side products which are environmentally benign and easy to remove via an aqueous work-up; 1-bromo-1-propene currently has no known severe health hazards. The facile method described below is safe and technically easy to perform. The high yield and reproducibility of this procedure make it a practical alternative to accessing multi-gram quantities of this important compound.
Procedure
An oven-dried 3 L round-bottom flask equipped with a magnetic stirring bar, a rubber septum, an electronic thermometer and flushed with nitrogen was charged with (Z/E)-1-bromo-1-propene (81.5 mL, 952 mmol),§ 1 L of anhydrous tetrahydrofuran (THF)¶ and maintained under a nitrogen atmosphere. The flask was cooled to −78 °C with a dry ice-acetone bath, and after 10 minutes n-butyllithium 2.5 M (800 mL, 2.00 mol)|| was added slowly to maintain the internal temperature below −60 °C. Following the end of the addition, the off-white mixture was stirred for 1.5 hours at −78 °C before methyl chloroformate (96.0 mL, 1.24 mol)** was added dropwise over 40 minutes in order to maintain the internal temperature between −70 °C and −60 °C. The mixture was stirred for 2 hours at −78 °C before the cooling bath was removed allowing the mixture to warm to room temperature over 1.5 hours. 500 mL of water were subsequently added and the organic layer was decanted. The aqueous fraction was extracted twice with 400 mL of diethyl ether and the combined organic fractions were dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure (40 °C, 100 mbar)†† to afford an orange brown liquid.‡‡ After distillation through a 5 cm long vigreux column (Bp = 64–67 °C, 60 mbar)§§ methyl but-2-ynoate (74.7 g, 80%)¶¶ was obtained as a colourless oil.||||

The elemental analysis and the spectral analysis of the product are as follows: Anal. Calcd for C5H6O2: C, 61.22; H, 6.16. Found: C, 61.12; H, 6.00%. 1H NMR (CDCl3, 500 MHz) δH 3.76 (s, 3H, CH3–O), 1.99 (s, 3H, CH3–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) ). 13C NMR (CDCl3, 125 MHz) δC 154.2 (C–CO2Me), 85.7 (CH3–C), 72.1 (CC–CO2Me), 52.5 (CH3–O), 3.7 (CH3–C). IR (film)/cm−1νmax 2957, 2247, 1714, 1436, 1263, 1075.
). 13C NMR (CDCl3, 125 MHz) δC 154.2 (C–CO2Me), 85.7 (CH3–C), 72.1 (CC–CO2Me), 52.5 (CH3–O), 3.7 (CH3–C). IR (film)/cm−1νmax 2957, 2247, 1714, 1436, 1263, 1075.
References
- For recent uses of 3 see: (a) G. Liu, Y. Shen, Z. Zhou and X. Lu, Angew. Chem., Int. Ed., 2013, 52, 6033 CrossRef CAS PubMed; (b) R. S. Foster, H. Adams, H. Jakobi and J. P. A. Harrity, J. Org. Chem., 2013, 78, 4049 CrossRef CAS PubMed; (c) Y. Liu, G. Wu and Y. Cui, Appl. Organomet. Chem., 2013, 27, 206 CrossRef CAS; (d) P. A. Wender, A. B. Lesser and L. E. Sirois, Angew. Chem., Int. Ed., 2012, 51, 2736 CrossRef CAS PubMed; (e) M. Onoe, K. Baba, Y. Kim, Y. Kita, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2012, 134, 19477 CrossRef CAS PubMed; (f) B. M. Trost, B. R. Taft, J. T. Masters and J.-P. Lumb, J. Am. Chem. Soc., 2011, 133, 8502 CrossRef CAS PubMed; (g) I. N. Michaelides, B. Darses and D. J. Dixon, Org. Lett., 2011, 13, 664 CrossRef CAS PubMed; (h) J. Burghart, A. Sorg and R. Brückner, Chem.–Eur. J., 2011, 17, 6469 CrossRef CAS PubMed; (i) H.-S. Yeom, E. So and S. Shin, Chem.–Eur. J., 2011, 17, 1764 CrossRef CAS PubMed; (j) P. A. Evans, J. R. Sawyer and P. A. Inglesby, Angew. Chem., Int. Ed., 2010, 49, 5746 CrossRef CAS PubMed.
- (a) J. Burghart and R. Brückner, Angew. Chem., Int. Ed., 2008, 47, 7664 CrossRef CAS PubMed; (b) T. Olpp and R. Brückner, Angew. Chem., Int. Ed., 2006, 45, 4023 CrossRef CAS PubMed; (c) M. A. Arnold, K. A. Day, S. G. Durón and D. Y. Gin, J. Am. Chem. Soc., 2006, 128, 13255 CrossRef CAS PubMed; (d) C. T. Brain, A. Chen, A. Nelson, N. Tanikkul and E. J. Thomas, Tetrahedron Lett., 2001, 42, 1247 CrossRef CAS; (e) G. B. Dudley, K. S. Takaki, D. D. Cha and R. L. Danheiser, Org. Lett., 2000, 2, 3407 CrossRef CAS PubMed.
- (a) Y. Qian, M. Hamilton, A. Sidduri, S. Gabriel, Y. Ren, R. Peng, R. Kondru, A. Narayanan, T. Truitt, R. Hamid, Y. Chen, L. Zhang, A. J. Fretland, R. A. Sanchez, K.-C. Chang, M. Lucas, R. C. Schoenfeld, D. Laine, M. E. Fuentes, C. S. Stevenson and D. C. Budd, J. Med. Chem., 2012, 55, 7920 CrossRef CAS PubMed; (b) J. R. Walker, S. C. Rothman and C. D. Poulter, J. Org. Chem., 2008, 73, 726 CrossRef CAS PubMed; (c) R. Lira, A. X. Xiang, T. Doundoulakis, W. T. Biller, K. A. Agrios, K. B. Simonsen, S. E. Webber, W. Sisson, R. M. Aust, A. M. Shah, R. E. Showalter, V. N. Banh, K. R. Steffy and J. R. Appleman, Bioorg. Med. Chem. Lett., 2007, 17, 6797 CrossRef CAS PubMed; (d) B. Cimetière, T. Dubuffet, O. Muller, J.-J. Descombes, S. Simonet, M. Laubie, T. J. Verbeuren and G. Lavielle, Bioorg. Med. Chem. Lett., 1998, 8, 1375 CrossRef; (e) K. Tatsuta, T. Yoshimoto and H. Gunji, J. Antibiot., 1997, 50, 289 CrossRef CAS; (f) J. W. Coe, C. R. Hawes and P. Towers, J. Labelled Compd. Radiopharm., 1995, 36, 587 CrossRef CAS; (g) G. Johnson, J. T. Drummond, P. A. Boxer and R. F. Bruns, J. Med. Chem., 1992, 35, 233 CrossRef CAS.
- (a) B. Kang, P. Jakubec and D. J. Dixon, Nat. Prod. Rep. 10.1039/C3NP70115H; (b) B. Darses, I. N. Michaelides, F. Sladojevich, J. W. Ward, P. R. Rzepa and D. J. Dixon, Org. Lett., 2012, 14, 1684 CrossRef CAS PubMed; (c) F. Sladojevich, I. N. Michaelides, B. Darses, J. W. Ward and D. J. Dixon, Org. Lett., 2011, 13, 5132 CrossRef CAS PubMed.
- R. B. Boers, Y. P. Randulfe, H. N. S. van der Haas, M. van Rossum-Baan and J. Lugtenburg, Eur. J. Org. Chem., 2002, 2094 CrossRef CAS.
- R. M. Moriarty, R. K. Vaid, V. T. Ravikumar, T. E. Hopkins and P. Farid, Tetrahedron, 1989, 45, 1605 CrossRef CAS.
- E. C. Taylor, R. L. Robey and A. McKillop, Angew. Chem., Int. Ed. Engl., 1972, 11, 48 CrossRef CAS.
- B. Myrboh, H. Ila and H. Junjappa, Synthesis, 1982, 1100 CrossRef CAS.
- A. Viale, D. Santelia, R. Napolitano, R. Gobetto, W. Dastrù and S. Aime, Eur. J. Inorg. Chem., 2008, 4348 CrossRef CAS.
- L. Brandsma and H. D. Verkruijsse, Synthesis, 1990, 984 CrossRef CAS.
- 1-Bromopropyne is oxygen sensitive and can spontaneously ignite upon exposure to air (see ref. 10).
- (a) T. T. Zung, L. G. Bruk, O. N. Temkin and A. V. Malashkevich, Mendeleev Commun., 1995, 5, 3 CrossRef PubMed. For an earlier related synthesis of 3 from propyne using a PdCl2–CuCl system see: (b) T. T. Zung, L. G. Bruk and O. N. Temkin, Mendeleev Commun., 1994, 4, 2 CrossRef PubMed. For a 19% yielding direct oxidative methoxycarbonylation of propyne using PdCl2–CuCl2 catalysis see: (c) A. G. Mal’kina, R. N. Kudyakova, V. V. Nosyreva, A. V. Afonin and B. A. Trofimov, Russ. J. Org. Chem., 2002, 38, 1088 CrossRef.
- (a) D. Toussaint and J. Suffert, Org. Synth., 1999, 76, 214 CAS. For a single report of an ester synthesis using a similar method see: (b) A. Otaka, E. Mitsuyama, T. Kinoshita, H. Tamamura and N. Fujii, J. Org. Chem., 2000, 65, 4888 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3qo00072a |
| ‡ Current address: Institut de Chimie des Substances Naturelles, CNRS UPR2301, Centre de Recherche de Gif, Bâtiment 27, 1 Avenue de la Terrasse, 91198 Gif-sur-Yvette Cedex, France. |
| § 1-Bromo-1-propene (cis and trans, 98%) (CAS 590-14-7) was purchased from Sigma-Aldrich Chemical Company, Inc. (Cat. no. B78203) and used as received. |
| ¶ Anhydrous tetrahydrofuran was obtained by filtration through activated alumina (powder ∼150 mesh, pore size 58 Å, basic, Sigma-Aldrich) columns. |
| || n-Butyllithium, 2.5 M solution in hexanes, AcroSeal was purchased from Acros Organics and used as received. |
| ** Methyl chloroformate (99%) (CAS 79-22-1) was purchased from Sigma-Aldrich Chemical Company, Inc. (Cat. no. M35304) and used as received. |
| †† Employing these conditions for concentrating in vacuo minimized product loss on the rotary evaporator. |
| ‡‡ Depending on the scale, the colour of the crude product may change from pale yellow to brown; however, this has no significant effect on the yield. |
| §§ Towards the end of the distillation, the residue was transferred to a smaller flask and the distillation was continued to recover the maximum amount of product. |
| ¶¶ Similar results were obtained starting with 40 mL and 10 mL of 1-bromo-1-propene; methyl but-2-ynoate was afforded in 84% and 77% yields respectively. |
| |||| The product is stable at −20 °C for at least three months. |
| This journal is © the Partner Organisations 2014 |